シンバイオ製薬株式会社 2019年12月期第2四半期決算説明会
シンバイオ製薬、輸入元の品質トラブルで業績予想修正 中長期的な黒字化にむけ経営資源を集中
アステラス製薬の連結子会社アステラス・ドイッチランド GmbH製造品 トレアキシン®凍結乾燥注射剤 品質問題

吉田文紀氏:2019年8月7日の午後に、業績見通しの修正をしておりますので、その補足説明をさせていただきたいと思います。
今期の業績予想を大幅に修正いたしましたが、中期経営計画に対しての影響としては、おそらく今期、場合によっては来期に及ぶ可能性はあるかと思います。
今回、私どもも考えられないような「トレアキシン®」の品質問題が起こっております。2018年の暮れからすでに25ミリグラムのバイアル品質問題が生じていたため、100ミリグラムを25ミリグラムの代替として市場に供給していたわけです。
ところが、2019年4月、5月あたりから、100ミリグラムバイアルについてもかなりの品質問題が出てきています。
異物混入についてご説明いたします。スライド3ページの写真で黄色い丸で囲った黒い点は、2ミリメートル×1.2ミリメートル(の異物)です。私が眼鏡を外してでも、この黒い点ははっきりと見えますが、こういったものが混入しており、いくつかのバッチにおいて確認ができました。当然市場に流通させるわけにはいきません。
さらに4月から6月の間に入ってきたバッチのなかから、外観不良といわれるものがかなり多く出てきております。(検出率が)3パーセントだったものが9パーセントになり、40パーセントを超し、医薬品としては極めて大きな問題であるため、現在は輸入を抑えています。
みなさんご存知かどうかわかりませんが、FDAが定めているGMPの規定の1つに、CAPAというのがございます。GMPはいわゆる適正な製造規範ということで、すべての製薬会社が守らなければならない1つの規律であるわけですが、それを逸脱した問題が出てきた時には、CAPAを設置して、徹底して再発を抑えなければならないというものです。
CAPAというのは、最初のCAはCorrective Action、是正措置です。PAはPreventive ActionでCAPAと呼んでいますが、これはすでにGMPの規範のなかに定められているものであります。
異物混入や40パーセントを超す外観不良などが起こった時には、企業としては当然このGMPに照らし合わせてCAPAをとらなくてはいけないというのが、FDAが定めている適正製造規範でありまして、これは当然日本の企業にも当てはまります。
欧州の経営陣では対応が遅く、なかなか話が通じないということもあり、私どもは現在、アステラス・ドイッチランドだけではなくアステラスの本社を含めて対応をお願いしているところであります。
CAPAの一番重要で求められるところは、異物の根本的な原因究明をすることです。それに対して、今度はCorrective Actionということで、是正措置を設けなければなりません。そして、このような不良品が高い比率で起こらないように、Preventiveに予防措置をとらなくてはいけないと定められております。
アステラスと私どもの間にあるライセンス契約及び供給契約の不履行ではないかということで、こういった問題が二度と起こらないように、CAPAを設置してほしいとかなり強力にお願いしております。
しかし、患者さんには絶対に供給を切らすわけにはいきません。アステラスが使っている2つの工場のうちベルギーにある1社をC社と呼び、ドイツにあるもう1社はO社と呼んでいますが、一貫してO社(の製造品)に(品質の)問題が生じているため、C社の製造品をシンバイオに回してほしいということをお願いしております。
2019年12月期 通期業績予想の修正
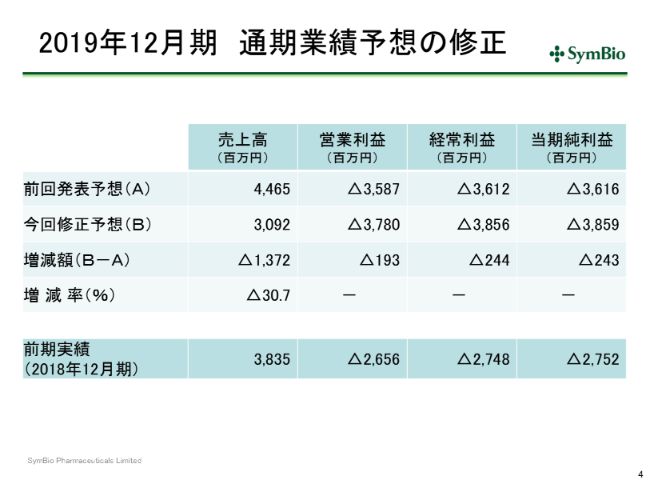
こういう状況ですが、スライドにあるとおり、今期の私どもの当初予定していた44億6,500万円から、30パーセントダウンの30億9,200万円への修正ということになります。これは前期比で20パーセントほどのダウンとなってきます。
当期純利益につきましても、経費をかなり抑えましたので、当初の予定の36億1,600万円の損失から、38億5,900万円の損失となっております。ネットで2億4,300万円の損失の増加ということで、抑えることはできてはおりますが、本来ならばこういった余計な損失は生ずるべきではなかったと思っております。
中期経営計画の修正
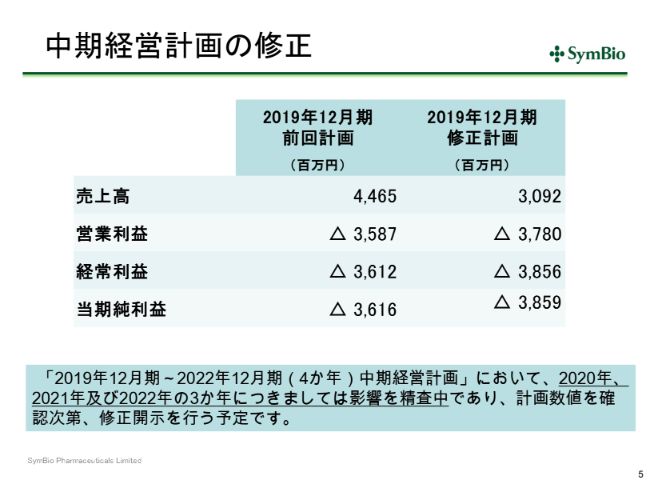
当然これは中期経営計画にも反映されるべきでありますので、スライド5ページの表左側の数字を右側のように修正しております。
今後どうなるのかというご心配はあると思いますが、現在精査中であります。アステラスのCAPAに基づく安定供給が回復するのがいつかによるかと思いますが、いずれにせよ市場への供給は切らさないという方針でアステラスと協議を続けているところであります。
のちほど詳しくお話し申し上げますが、開発が非常に順調に進んでおりますので、2021年になるとおそらく間違いなく液剤に切り替わっていくと考えております。
中期経営計画で掲げている2021年黒字化のために全力投球

私どもとしては、こういった5つの問題はありながらも、まずは中期経営計画で掲げております2021年の黒字化のために着実に実行していくことに、現在全力投球しております。
中長期業績 vs. 今期業績

品質問題により当期業績の損失は拡大します。突然大雨になった、台風が来たといったような状況のなかで対応を迫られているわけですが、中長期的にはパイプラインが着実に進展しており、自社販売体制構築も非常に順調に進んでいると考えております。
詳細については控えさせていただきますが、新規のライセンス候補が出てきておりまして、基本条件については先月合意に至っております。
今期の業績は非常に難しい状況ではありますが、中長期的にはほとんど変わりないと考えていただいていいかと思います。
2019年度上半期 損益計算書
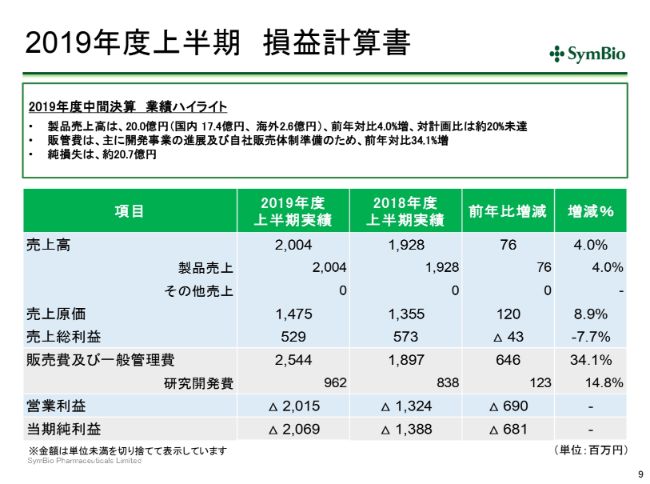
中間決算概要に入ってまいります。今の品質問題を反映させることによって、この1ー6月期は国内で17.4億円、海外で2.6億円ということで、合わせて約20億円となり、前年比で4パーセント増ですが、対計画比は約20パーセントの未達ということになってまいります。
経費は予定どおり開発が進んでおりますので増加しており、営業体制構築ということでインフラにも投資をしておりますので、34.1パーセントの増となっております。
2019年度上半期末 貸借対照表
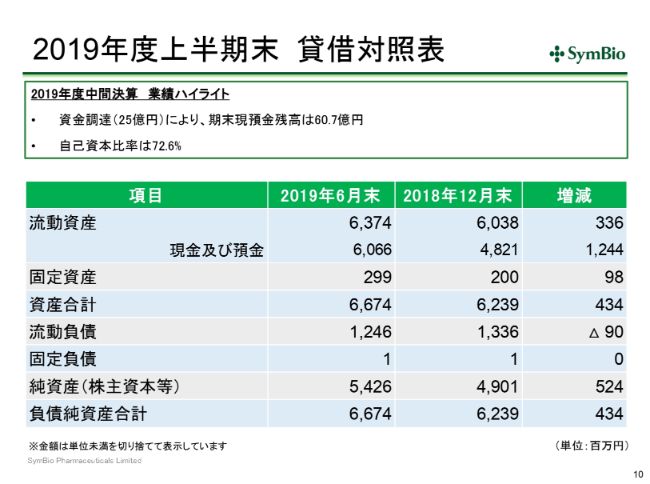
3ー6月で資金調達を行って、約25億円調達しておりますので、6月末の現金残高は60.7億円になっております。
2019年度 通期業績予想
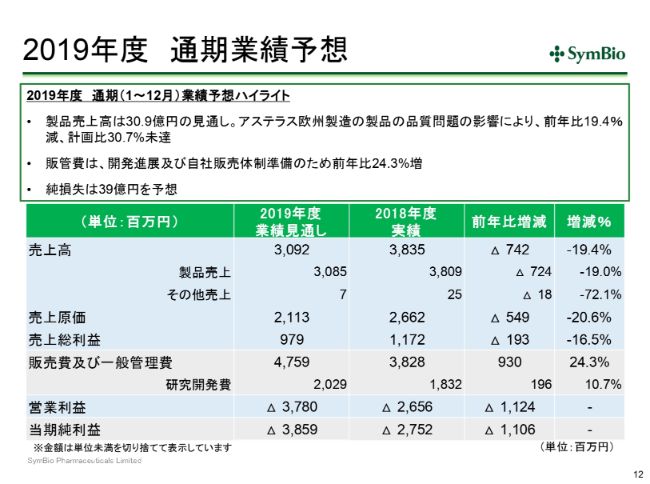
通期見通しは先ほどすでに一部ご紹介しておりますが、今期の売上見通しは約31億円と、前年比で約20パーセントダウンで、対計画比で30パーセント未達ということになってまいります。
販管費は予定どおり投資を進めてまいりますので、前年比で24パーセント増となってまいります。その結果、今期の純損失は39億円ということであります。
2019年度上半期のハイライト

事業の進捗ですが、非常に順調に来ております。国内の薬価ベースの売上は、前年とほぼ同じぐらいの水準で動いております。私どものMRは、9月末に3名追加して20名体制になりますので、生産性をもってくる今期末または来期の第1四半期には、おそらくこの成長率が上向いてくるのではないかと期待しております。
7月1日からこの17名が全国に配置されておりまして、全国のエーザイさんの各営業所のMRの方たちと共同で、プロモーションが進んでおります。流通、物流、営業などのインフラも順調に進展してきております。
パイプラインの詳細については、次のスライドでお話しいたしますが、先ほど申し上げたように、新規ライセンス導入については基本条件が合意に至っているということです。
約25兆円の資金調達が完了して、ザ・メディシンズ・カンパニーとの仲裁も、6月にニューヨークにおいてかなり進捗が見られております。現在、3分の2ぐらいのところまで来ていると考えておりますので、年末までにはこの仲裁は終わって、結論が出ると考えております。
7月1日から全国展開を開始
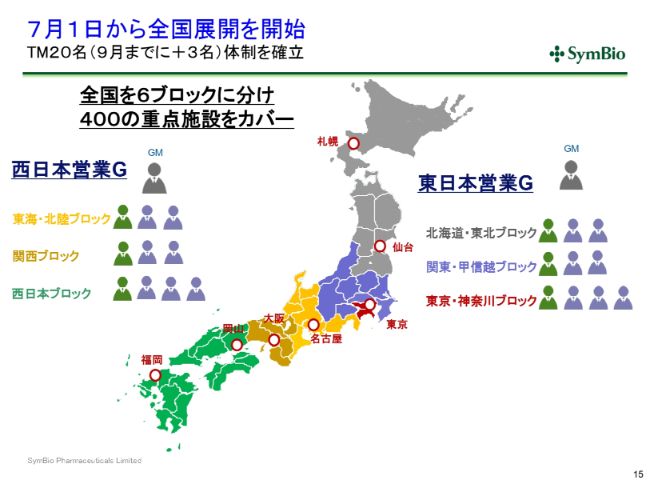
スライドのようなかたちで、20名が全国に配置されてまいります。すでにこのうちの7割が全国に配置されていると考えてよろしいかと思います。将来的には7ブロックに分けるという考え方でおりますが、現時点ではまず6ブロックで400重点施設をカバーしていくことを考えております。
BR療法は標準療法としてRーCHOPを抜き市場浸透率No.1
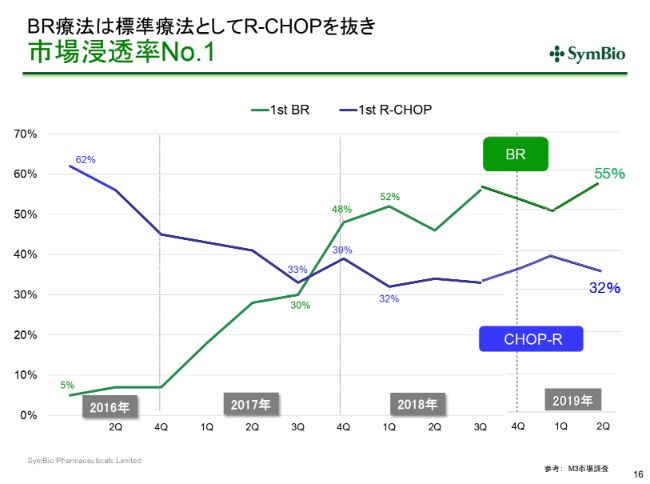
(BR療法は)すでに従来の標準療法RーCHOPを抜き去って、若干フラットになったように見えますが、今期末から来期にかけて、60-70パーセントにまで上向いていく可能性があるかと考えております。
私どもで採用した「トレアキシン®」マネージャー20名に加え、おそらくあと10名ほど自社のMRを追加採用するということになるかと考えております。その30名が生産性をもち始めた時に、70パーセントに向かって(BR療法の)シェアが伸びていくと期待しているわけです。
2019年度上半期のハイライト トレアキシン® 開発パイプラインの着実な進展
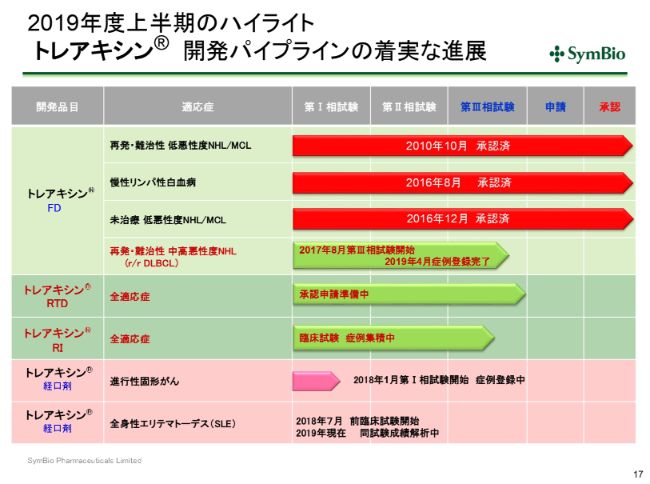
パイプラインにつきましては、3つの適用承認をとっております。この再発・難治性DLBCLについてはすでに症例集積を終えておりまして、現在承認申請に向けて準備を開始しております。
「トレアキシン®」のRTDは液剤ですが、これも一応予定どおり9月、10月には承認申請に至ると考えていただいてよろしいかと思います。そこから約1年で承認に至ると予定しております。
RIにつきましては、36症例を集積するということで、現在も鋭意活動しています。すでに半分近く症例集積が進んでおりますので、おそらく来期の第1四半期には、36症例の集積が完了するのではないかと考えております。
「トレアキシン®」の経口剤につきましては症例集積をしており、現在は16症例が入っております。そのなかで、サードライン、フォースラインということで他の抗がん剤によってかなりの治療を受けてこられた患者さんのアクティビティが若干乱れてきています。そのため今後はがん種を絞るプロセスに入ってまいりますが、この判断につきましては今期末になるかと思います。
SLEにつきましては、現在前臨床試験をほぼ終えまして、成績について解析中です。
新剤型×適応拡大によるトレアキシン®の事業価値の創造
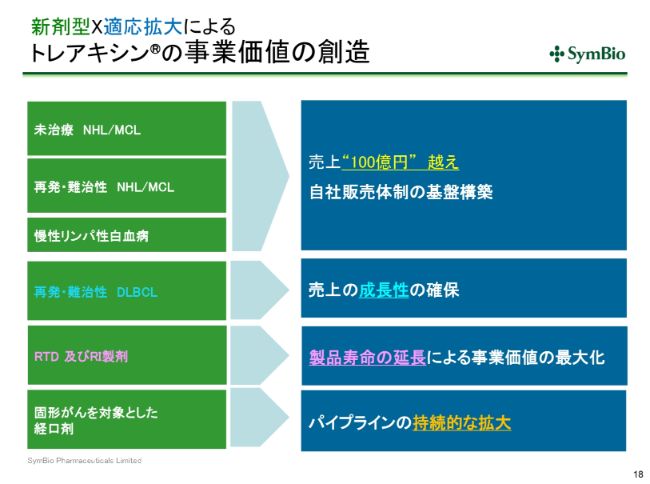
シンバイオの事業は、多数の適応症と、新しい剤型のなかで事業価値を創造していこうというものです。すでに承認がとれているこの3つの適応症については、自社販売体制構築をするなかで100億円を超すといった使命を持っていますが、さらに売上を伸ばして成長性を確保していくため、2021年にDLBCLの承認をとってまいりたいと考えています。
何回かすでにお話ししておりますが、液剤につきましては2031年まで特許により保護された期間があるため、製品寿命を大幅に延長できるという大きなメリットがございます。
固形がんは、経口剤によって固形がんを対象として、さらなるパイプラインの拡大をしていこうと考えています。
トレアキシン® 再発・難治性DLBCL第Ⅲ相臨床試験 症例登録を完了し、承認申請に向けて準備開始

「トレアキシン®」のDLBCLの第Ⅲ相試験につきましては、40症例で登録を完了いたしまして、現在は承認申請に向けて準備を開始しております。
私どもの場合、BRということで、「トレアキシン®」の用量が120ミリグラムというものを使っております。ロッシュとジェネンテックが開発した「Polatuzumab+BR」という新たな療法がありますが、そちらの治療方法はBの用量が90ミリグラムでありますので、若干比較にはならないものの、いずれにせよもう1つの新たな治療の選択肢が生まれつつあるということです。
NCCN Guidelines Version 1.2012 Diffuse Large B-Cell Lymphoma

BRについては、2012年のNCCNのガイドラインで推奨された治療方法となっており、欧米においてはすでに標準的に使われている治療方法と言えます。
私どもは2021年に承認をとるため、欧米に遅れること10年とは言いませんが、8~9年という長い時間をかけて、ようやく日本においてBRが使われるようになると期待しております。
2019年6月10日、Polatuzumab+BR(PーBR療法) 再発難治性DLBCLで迅速承認をUS FDAから取得

さきほど申し上げた、ジェネンテックとロッシュによって開発され、日本では中外製薬が開発中の「Polatuzumab+BR」は、ベンダムスチンとリツキシマブの併用です。
3剤併用のこの治療方法が、2019年の6月10日にUS FDAから迅速承認を得ておりまして、CR率とORRという全奏効率は、BRが120ミリグラムの時とそれほど変わらない結果にはなっておりますが、患者さんにとっては新たな選択肢となり、いいことであると考えております。
私どもにおいても、中外製薬さんによってこの「Polatuzumab+BR」が使えるようになっていきます。販売されて先生方がお使いになるということは、競争ではないと考えておりますし、むしろ患者対象が広がっていく可能性も十分あるかと期待はしております。
再発難治性DLBCL市場は「空白の治療領域」...患者さんの切実な声

DLBCLの領域については患者会の代表の天野さんがおっしゃっていますが、なにしろなにもないということのなかで、多剤併用療法というものが使われています。
これは5つぐらいの非常に強い抗がん剤の組み合わせとなりますので、高齢者にとっては非常につらい治療方法ですが、BRまたは「Polatuzumab+BR」が承認されて使えるようになりますと、おそらく流れは一気にそちらに切り替わっていくものと考えております。
2021年度中にRTD製剤へ 2022年度末にはRI製剤への100%切替えを目指します
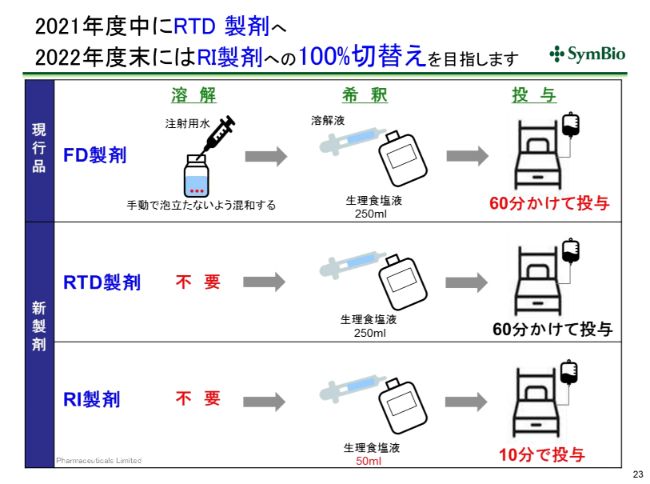
液剤につきましては、現在の凍結乾燥品は溶解に非常に手間暇がかかりますので、手間暇が省けるRTDで置き換えていきます。
あとのステップはほぼこの凍結乾燥品と同じプロセスになりますが、RIになりますと、50ミリグラムで希釈をするだけで済みます。投与も10分間ですので、患者さんにとっても医療従事者にとっても、負担の軽減に繋がるため、私どもとしては1日も早くこのRIに置き換えていく努力をしていきたいと考えております。
米国市場において液剤RI(BENDEKA® )への切替えは発売後1年以内に90%以上を達成
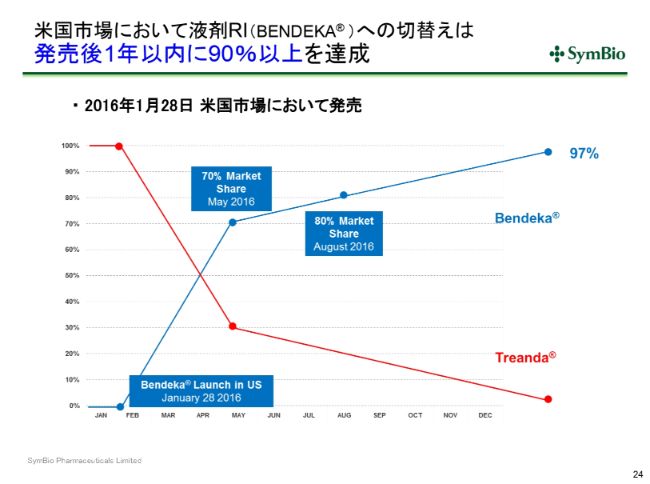
これはアメリカの置き換えのペースですが、アメリカではRTDを実施せず、一気に「Bendeka®」という液剤のRIにもっていきました。「Treanda®」というのが凍結乾燥品です。
2016年1月28日に発売をしたあと、同年4月末~5月にかけて、約3~4ヶ月で7割がRIに切り替わっています。2016年末までには90パーセント、2017年目の終わりには97パーセントという切り替えのスピードですので、おそらく日本においてもこのようなかたちになるかと思っております。
トレアキシン®事業戦略 標準療法の強みをフルに活かした展開
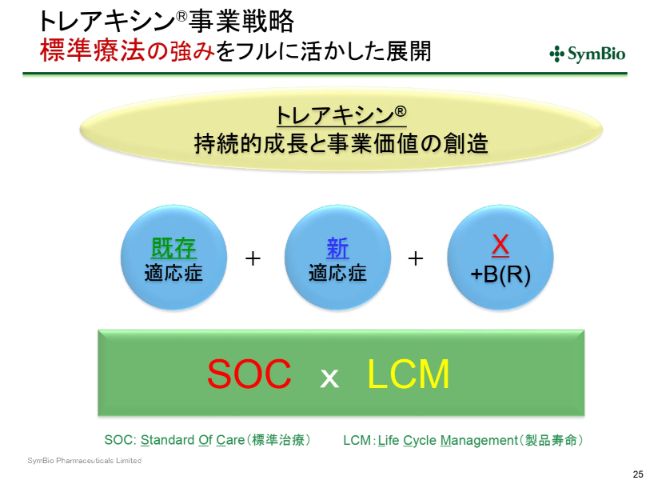
製品寿命が2031年まで大幅に延長できるようになることは、すでに標準療法としてこの3つの適応症で使われ、今度はDLBCLにおいても使われていくことになってきます。10年以上の時間のなかで、「X+B(R)」というものが、先ほどの「Polatuzumab+BR」のようにして、新たに生み出される可能性は十分あると考えております。
2030年に向けて悪性リンパ腫の患者数 年平均2~3%で増加する見通し
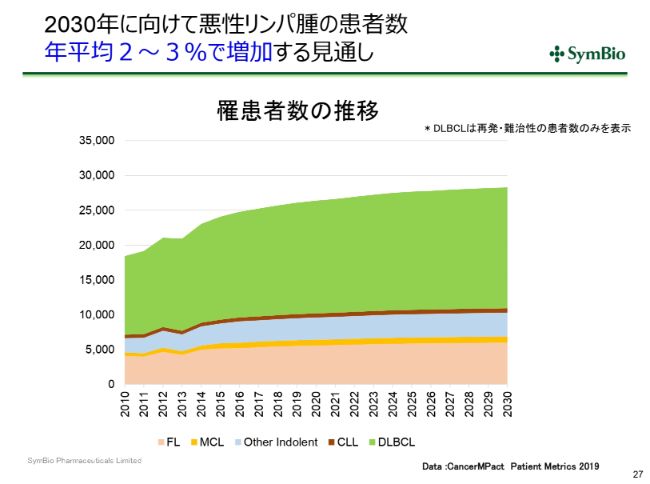
悪性リンパ腫は高齢者の疾患であり、とくに高齢化が進んでいく日本においてはだいたい年に2~3パーセントで患者さんの数が増えていくであろうと考えられます。
Eagle社は現在新たに特許を登録しようとしておりますが、場合によっては2023年(になる)ということです。ある意味では、この(悪性リンパ腫の)増加傾向が、液剤に切り替えることによって売上に結び付いてくるまでの時間を十分に与えてもらえるということになってくるわけです。
リゴセルチブ注射剤と経口剤 複数の適応症を対象として開発を推進
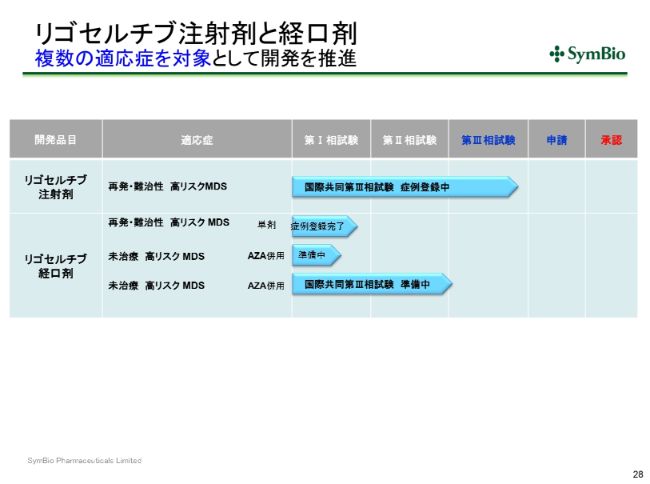
「リゴセルチブ」についてですが、注射剤についてはすでに44症例(集積されており)、目標は50症例ということです。
リゴセルチブ注射剤 HMA不応高リスクMDS対象国際共同第Ⅲ相試験

オンコノバ社も目標は360症例ですが、すでに300症例近く組み入れが終わっておりますので、あと半年ぐらいの間に、グローバルの症例集積も完了するのではないかと思っております。私どもも今期末には間違いなく50症例を完了しておきたいと考えております。
リゴセルチブ経口剤 第Ⅱ相臨床試験結果 2018年米国血液学会議(ASH)にて発表
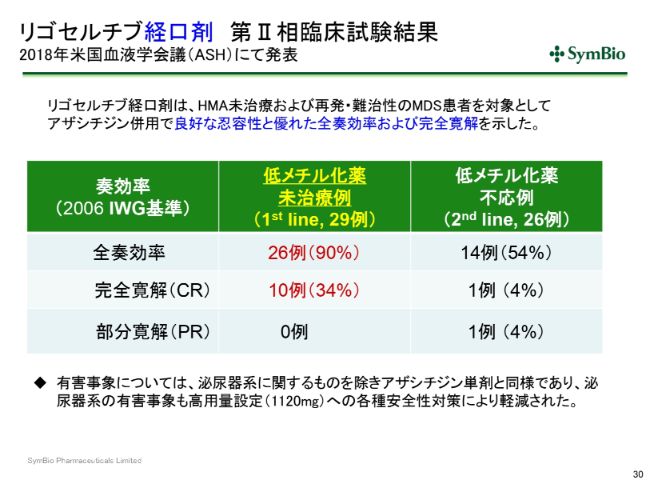
経口剤につきましては、「アザシチジン」との併用において、良好な忍容性と優れた全奏効率および完全寛解を示しておりまして、これを再現しようという第Ⅲ相試験が現在検討されております。
リゴセルチブ経口剤 未治療高リスクMDS対象 国際共同第Ⅲ相試験(AZA併用)開始準備中

FDAとSPAについて、現在オンコノバ社は協議中であり、おそらく2019年9月末にはFDAとの了解がとれるのではないかと考えております。
私どもとしては、オンコノバ社がこのSPAを取得したのち、できるだけ早く国際共同第Ⅲ相臨床試験に参加するという予定でおります。
中期経営計画 目標達成のために経営資源を集中

今後の予定ですが、先ほど申し上げたようにこの5つの目標については必達でありまして、今期、来期そして再来期と、この5つの目標達成のために、経営資源の約90パーセントを集中させていくことになるかと思います。
中期経営計画 収益モデルへの転換期
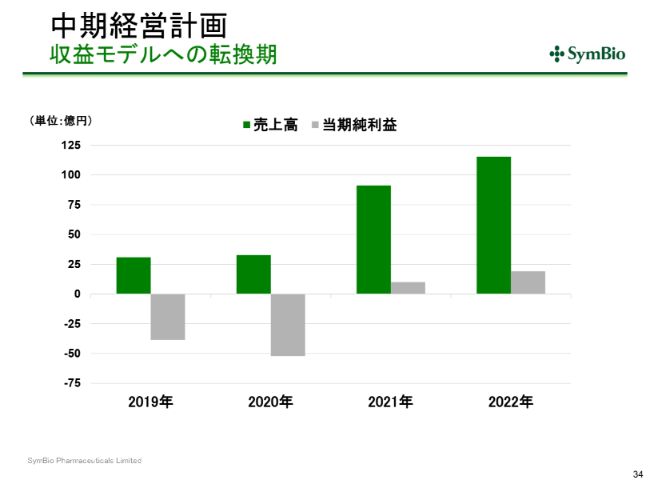
そうすることによって、収益モデルへの転換期が、S字カーブを描くことができると考えております。
経費投資の推移 血液領域専門プラットフォーム構築への投資
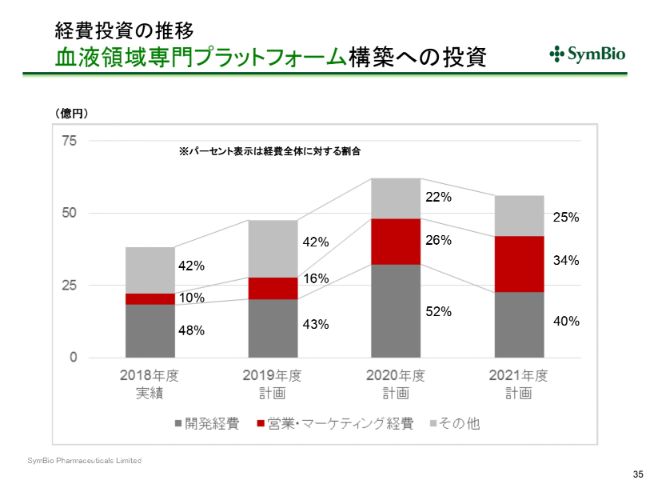
2018年には、営業・マーケティング経費は、すべての経費のなかの10パーセントを占めていたわけですが、2019年には16パーセント、来期には26パーセント、そして2021年には34パーセントと、全経費に占める営業・マーケティング経費が増えてまいります。
おそらく、開発経費はだいたい40パーセントで、年によって少しでこぼこするとは思いますが、今後はだいたいこのような感じで推移していくのではないかと思います。
ご覧になってわかりますように、これは経費の金額でのトータルで、2020年が若干のピークとなってまいります。資金を切らさないように十分な資金を持ち、2020年をしっかりと乗り越えることによって、S字カーブを描いてまいりたいと考えております。
しっかりとこのようなかたちでお示しすることができますと、いわゆる血液疾患をベースとするスペシャリティファーマができあがります。その時には、かなりの収益になってくると思いますので、今回新たに導入する新薬についても、今までのインフラをそのまま使え、上乗せできるという考えで、新薬の導入に踏み切ったわけです。
収益モデルへのトランスフォメーションのイメージ 収益性と成長性を兼ね備えた”第二のシンバイオ”の創業
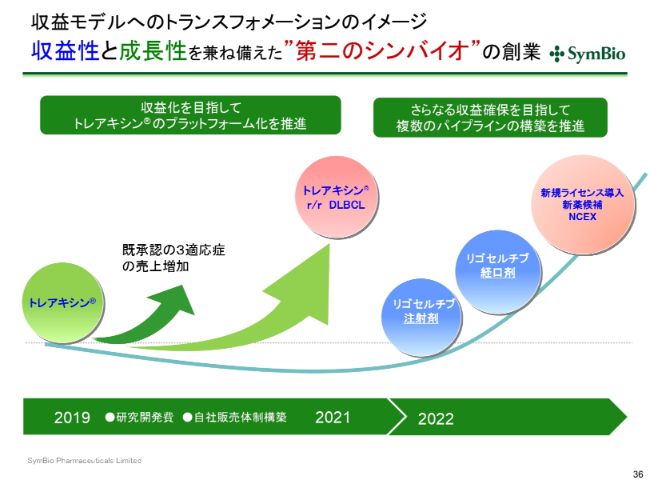
考え方としては、現在の3つの適応症でできるだけ早く100億円を達成し、「トレアキシン®」のDLBCLで上乗せし、200億円に向けて売上を伸ばしていきたいということです。
その間に「リゴセルチブ」の注射剤、経口剤といったものの承認を取り、まだ完了しておりませんが、導入したあかつきには、2025、2026年ごろにはこの3本目の柱が出てくると考えております。
“深化と進化”を続けるシンバイオのパイプライン戦略
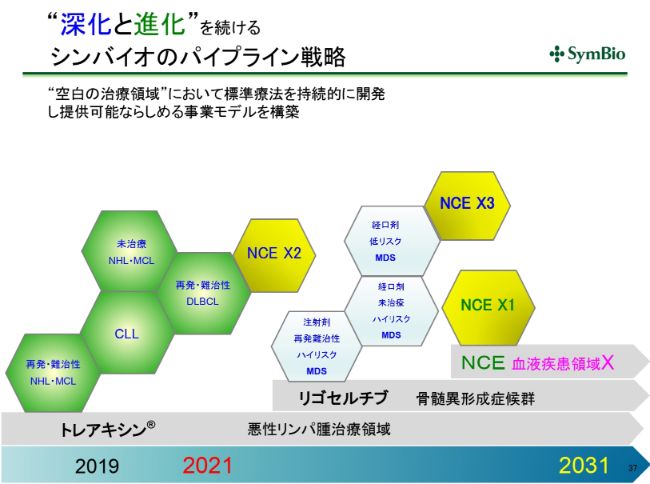
パイプラインに対する考え方は、悪性リンパ腫において標準療法をもっていることの強みをフルに活かしながら、さらにBRプラスで出てくるものをとっていくというのが1つだと思います。これを深化していく、すなわち深く掘り下げていくということです。
さらに、骨髄異形成症候群では現在多くの臨床試験が走っておりますので、私どもとしては「リゴセルチブ」プラス「アザシチジン」で標準療法を確立することができれば(と考えています)。
骨髄異形成症候群という疾患は非常にヘテロなものですから、「リゴセルチブ」プラス「アザシチジン」プラスもう1つ、というかたちで、そのヘテロの一つひとつを狙い撃ちしていくということも十分に考えられます。
実際にそういった開発をしているバイオベンチャーからアプローチもきております。「リゴセルチブ」プラス「アザシチジン」で適応症をとっておけば、今後開発されるものが「リゴセルチブ」プラス「アザシチジン」プラスXということになってくる可能性もあると思っております。
今回、導入することができれば、血液領域のなかの別のセグメントにおいて、新たな製品群を構築することができるようになってくるだろうと思います。
現在ここに重点を置いて導入を検討しておりますが、実際にNCEの候補が出てきていることも事実であります。「血液領域においてはシンバイオ」となるまでもっていくには、おそらく2021、2022年までかかると思います。ですが、おそらく非常に専門性の高くユニークな医薬品会社をつくることができると考えております。
シンバイオ製薬の企業使命と企業戦略

私どもの場合、常に医療ニーズがあるかどうか、「空白の治療領域」を埋めることができるかどうかによって導入をしてまいります。現在1つもいい薬がないような治療領域をターゲットに、私どもが新薬を導入します。これがうまくいけば今回は、グローバルな権利もとれるというところまで交渉は進んでおります。
難しい時期にはきておりますが、品質問題が将来に影響を及ぼさないように、ここ数ヶ月、アステラスと協議を続けていきたいと考えております。以上でございます。
新着ログ


「医薬品」のログ


