提供:株式会社キャンバス 2025年6月期決算説明
【QAあり】キャンバス、CBP501欧州臨床第3相試験申請・準備への注力継続 返還されたCBS9106の開発方針は今後追加検討
会社概要

加登住眞氏(以下、加登住):取締役CFOの加登住眞です。これより、2025年6月期決算説明会を開催します。よろしくお願いします。
当社は従業員15名の少数精鋭の体制で進めています。その他、会社概要等については、決算説明会にご参加の方々にはおなじみの内容かと思いますので省略します。詳細は当社Webサイトなどをご参照いただければと思います。
キャンバスの強み がん免疫に着目したパイプライン戦略

創薬バイオテック企業として、最初にパイプライン戦略をご紹介するのが恒例となっています。
「CBP501」は、当社が創業時から取り組んでいる抗がん剤候補化合物です。米国で臨床試験第2相まで終了し、第3相試験の実施を目指しましたが、第2b相の開始承認の取得となったことから、昨年より欧州での臨床第3相試験の開始に向けて準備・申請を進めています。
続いて、前回の決算説明会までStemline社に導出済みとご説明していた「CBS9106」については、第1相試験を完了するまで進められ、すでに開示しているとおり、その後すべての権利がキャンバスに戻されることとなりました。期末間際となる今年6月30日での公表となり、この決定自体がその直近でなされたため、今後の進行方向については現在検討中です。
前臨床試験の準備中である「CBT005」および「CBP-A08」については、水面下で次の開発パイプラインとなる可能性がある段階に進んでいます。
それでは、決算説明およびこの1年間の進捗について、河邊と私が順次ご説明します。まずは、開発パイプラインの状況について、代表取締役社長の河邊よりお話しします。
各パイプラインの歩み・現状・目標
河邊拓己氏(以下、河邊):代表取締役社長の河邊です。よろしくお願いします。
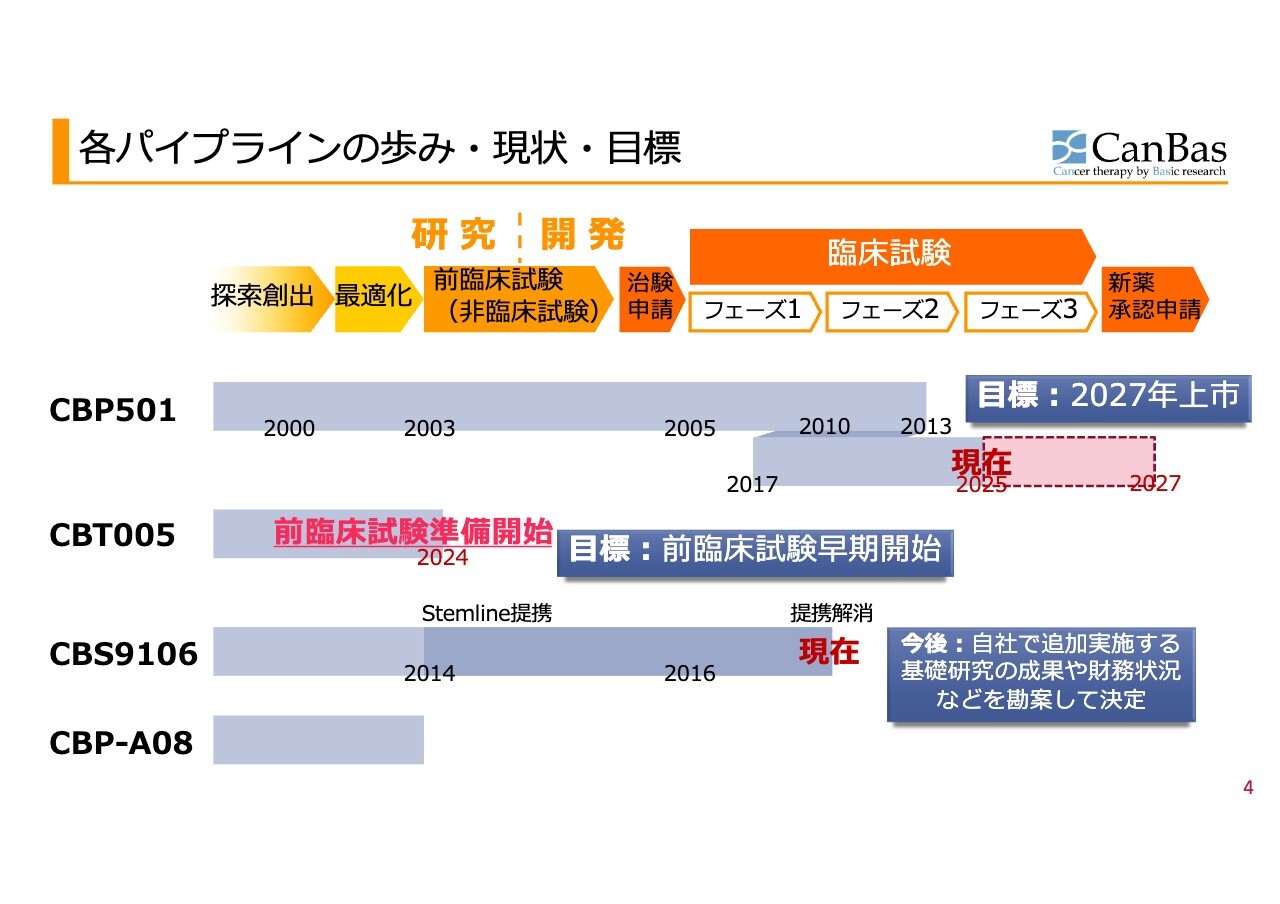
開発パイプラインの状況についてご説明します。各パイプラインの進捗はスライドのとおりです。
「CBP501」については、先ほど加登住も申し上げたとおり、創業当時からのパイプラインです。一時期、細胞障害性抗がん剤の全盛の時代の開発を進めてきましたが、紆余曲折を経て、免疫系抗がん剤を併用する臨床試験へと開発方針を大きく変更しました。
これは世の中が免疫系抗がん剤へシフトしたのとも同じタイミングとなり、当社にとってこの転換はたまたま幸運だったとも言えると思います。
「CBT005」については、現在まだ前臨床試験を開始する段階ですが、将来大きく成長してくれることを期待しています。
「CBS9106」については、先ほど加登住が申し上げたとおりです。
「CBP-A08」は「CBP501」の後継にあたります。
免疫系抗がん剤は一部の患者様に劇的に効く
最初のパイプラインである、免疫着火剤と呼ばれる「CBP501」についてもう少し詳しくご説明します。
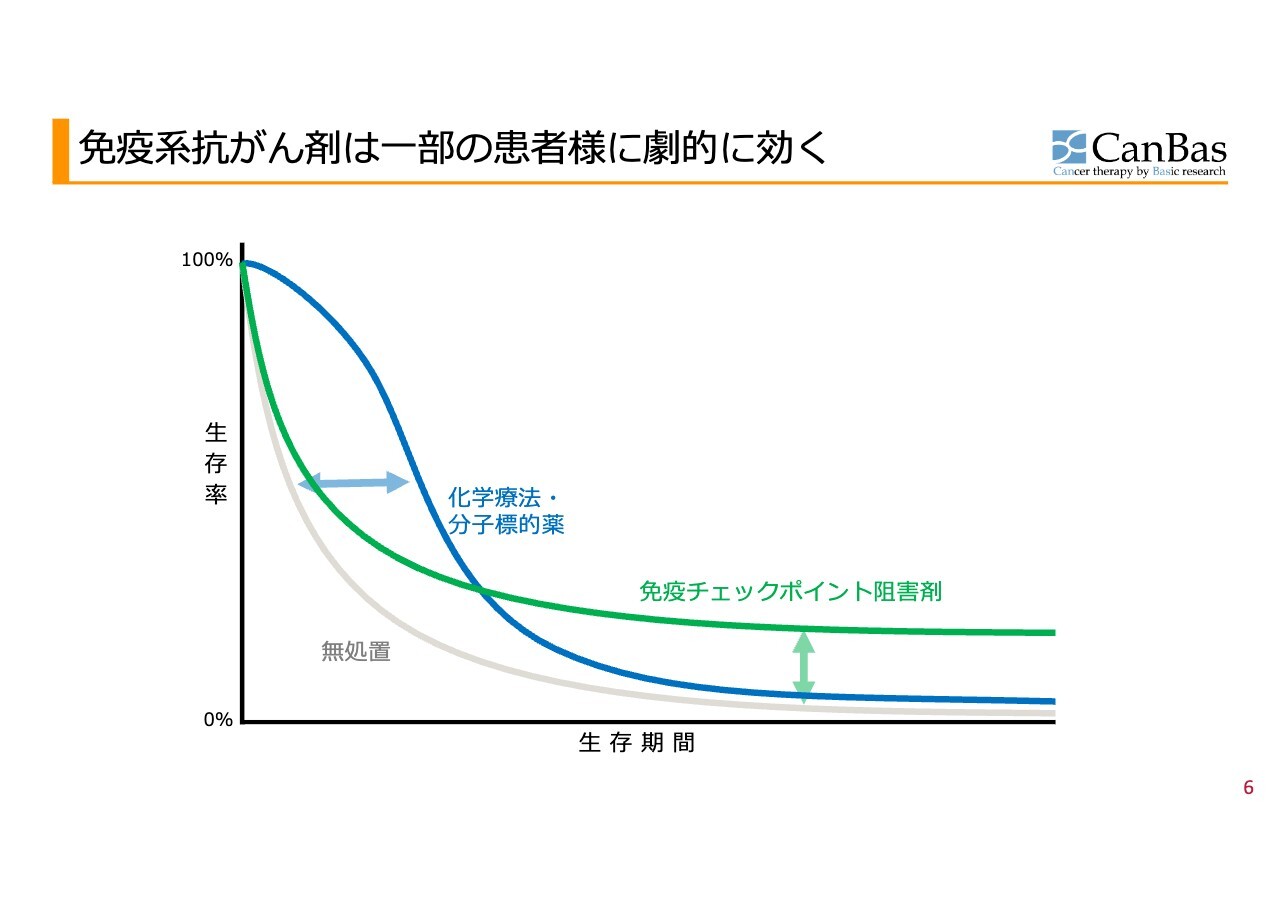
先ほど少し触れたように、時代が従来型抗がん剤の時代から大きく変わったということですが、スライドにはがん患者集団の生存期間と生存率を示したカプランマイヤー曲線を掲載しています。
この曲線は、横軸が集団の生存期間、縦軸が集団の何パーセントの方がその時点で生存しているかを表しています。
これまでの標準治療では、2014年頃まで主流であった従来型の細胞傷害性抗がん剤や分子標的薬の場合であれば、多くの対象患者さまに反応は見られるものの、長期的にはあまり効果が続かないことが課題でした。
一方で、免疫チェックポイント阻害剤は、効果が見られる患者さまが非常に限られるものの、効果が見られた場合は非常に長期にわたり作用が持続する特徴があります。このような変化により、抗がん剤の臨床開発が大きく変わる契機となりました。
免疫系抗がん剤の効きにくい「免疫コールド」ながん
そのような状況の中における、当社のパイプラインについてご説明します。
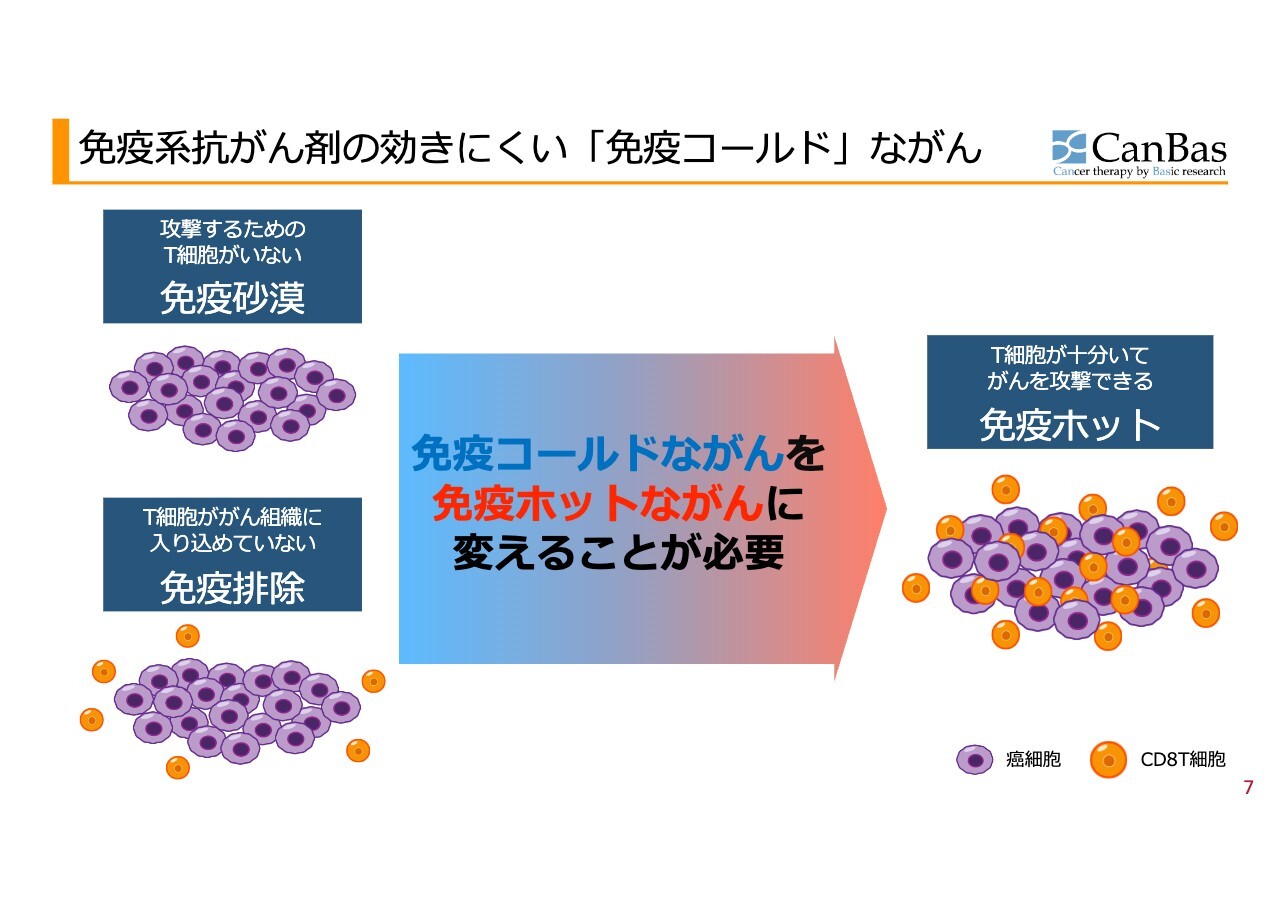
まず、先ほどお話ししましたように免疫系抗がん剤が一部の患者さまには効果があるものの大部分には効果がないという課題についてご説明します。
効果がないと考えられる患者さまにおいて、どのようなことが起きているかをスライド左に示しています。免疫系抗がん剤は、最終的にがんを攻撃するCD8T細胞、スライド内のオレンジ色で示された細胞が作用することで効果を発揮します。
しかし、一部の患者さまでは、がんを殺すためのT細胞がそもそもがん組織の近辺に存在しないために効果が発揮されない「免疫砂漠」の状態が見られます。また、T細胞自体は存在しても、それらががん組織に入りこむことができず、がん細胞に到達できていない「免疫排除」の状態があると考えられています。
このような患者さまには、現在注目を集めている免疫チェックポイント阻害剤「オプジーボ」などが効果を発揮しないことがわかっています。
そのため、これらの状態をまとめて「免疫コールドながん」と呼びますが、スライド右に示しているように、免疫細胞ががん組織内に存在する「免疫ホット」な状態へと変化させることが必要であるとの認識が広まっています。
CBP501の3剤併用で免疫系抗がん剤を効きやすくする
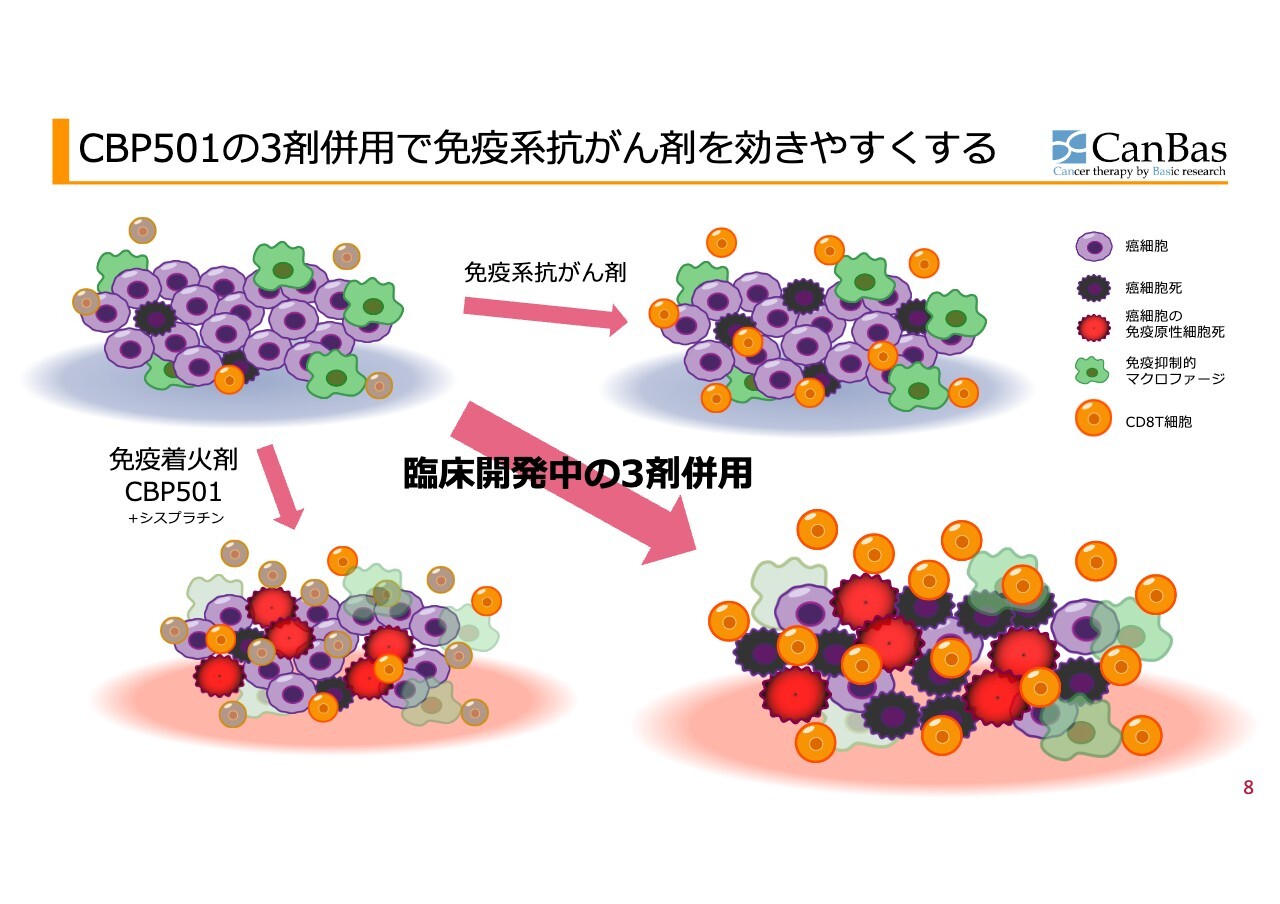
膵臓がんの場合について、もう少し詳しくモデルで示します。
スライド左上が膵臓がんのイメージ図です。紫色で示されたがん細胞に対し、がんを殺す細胞であるCD8T細胞については、元気な細胞は小さく丸いオレンジ色のものが1個だけで、あとはくすんだ色をしています。この状態は、ブレーキがかけられていることを表現しています。
さらに、免疫を抑制する免疫抑制系の細胞が多数存在します。ここでは代表例としてマクロファージのような細胞を緑色で表しています。
次に、紫色で表された癌細胞死は、濃い色のトゲトゲした形で表しています。がん細胞が死ぬ際には、アポトーシスという免疫反応を起こさないような細胞死が多く見られます。この状態が「免疫コールドな状態」と呼ばれ、膵臓がんがその代表例です。
ここで、スライド右上のように「オプジーボ」のような免疫系抗がん剤を使用すると、T細胞のブレーキが外れるため、CD8T細胞が活性化します。
しかし、CD8T細胞の数自体が少ないことに加え、免疫抑制系細胞や、アポトーシスによる免疫抑制の影響により、特に膵臓がんでは免疫チェックポイント抗体単体では効果が得られないことがすでに証明されています。
そこで、当社の免疫着火剤について説明します。このような状況の中で、スライド左下のように「CBP501」と「シスプラチン」を組み合わせることで、まず「シスプラチン」の作用を強化し、がん細胞を静かな死に方ではなく、免疫を活性化させるような派手な死に方へと導きます。これは図中に赤色で示しています。
さらに、2014年以降の研究で明らかになったことですが、「CBP501」には免疫抑制細胞の作用を抑制する効果があります。具体的には、免疫抑制マクロファージのサイトカイン放出を抑える作用が確認されています。
つまり、この免疫着火剤は、免疫抑制細胞を抑制することによりT細胞ががん細胞内へ浸入できるようにします。免疫抑制が解除されることに加え、赤い色で示した「免疫原性細胞死」という派手な死に方が誘導されることで、さらにT細胞ががん細胞内に入りやすくなります。
ここでいよいよ「オプジーボ」のような免疫チェックポイント抗体を加えることで、スライド右下のようにT細胞が多数活性化した、がん細胞を排除するように働く状態になると考えており、当社はこのようなことを目指しています。
また、マウスの実験においてはもちろん、臨床第1b相試験においてもこれに似た現象が起こっていると考えられるデータを得ています。
CBP501臨床第2相試験データ学会発表

そこで、臨床試験第2相試験を実施しました。直近の第2相試験に関する試験結果のまとめをお話しします。このデータは、臨床試験第2相の学会発表で報告したものです。
試験結果は、結果が出た時点でプレスリリースなどにより部分的に報告していましたが、学会発表や論文として公表されることが医学界では非常に重要です。今回の発表は学会でのポスター発表となり、その時点での試験結果をダイジェストとしてお伝えします。
主要評価項目とは、臨床試験を開始する前に「ここを見ますよ」として宣言し、その結果を確認する最も重要な指標です。この試験では、3ヶ月後の無増悪生存率を主要評価項目として設定しました。
具体的には、3ヶ月の時点で何パーセントの患者さまのがんが進行していないかを評価し、一定以上の割合を達成することが目標となっています。
第1群では、3剤併用療法で「CBP501」「シスプラチン」「オプジーボ」を用いました。「CBP501」は、当社が使用を予定している投与量で25mgです。
第2群では、「CBP501」の投与量のみを16mgに減らし、「CBP501」「シスプラチン」「オプジーボ」の組み合わせで試験を行いました。
そのほかに、薬剤の組み合わせの異なる2つの群を設定しました。
この試験の結果は非常に良好でした。本試験は、中間でいったん解析を行い、その結果を基に後半を実施するかどうかを判断する構造でしたが、第1ステージで非常に良い結果が得られました。
その結果、実際に患者さまたちを診ていた臨床医たちの会議である安全性モニタリング委員会で「これ以上やる必要はない」「次の試験に進みなさい」という方針が決まりました。
重要な点として、副次的評価項目である無増悪生存、客観的奏効率、病勢コントロール率、奏功期間などにおいても、主要な効果と同様に明確な状況証拠が得られたことが挙げられます。
最も重要なのは安全性です。結論として、当社と直接の利害関係を持たない医師たちによる発表では「CBP501」「シスプラチン」「オプジーボ」の併用療法は転移性膵臓がんの3次治療として忍容性のある安全性を示し、抗がん剤として極めて重要な安全性の許容範囲内であると評価されました。
また、3ヶ月の無増悪生存率、増悪生存期間および全生存期間において、持続的な奏功と臨床的に意義のある改善をもたらしたことが記載されています。
なお、「臨床的に意義のある改善」と記載することは、学会や論文において非常にハードルが高く、審査者が「そこまでは言えない」と、記載が認められない場合もよく見られます。したがって、これは極めて重要なポイントとなります。
CBP501臨床第2相試験データ論文発表
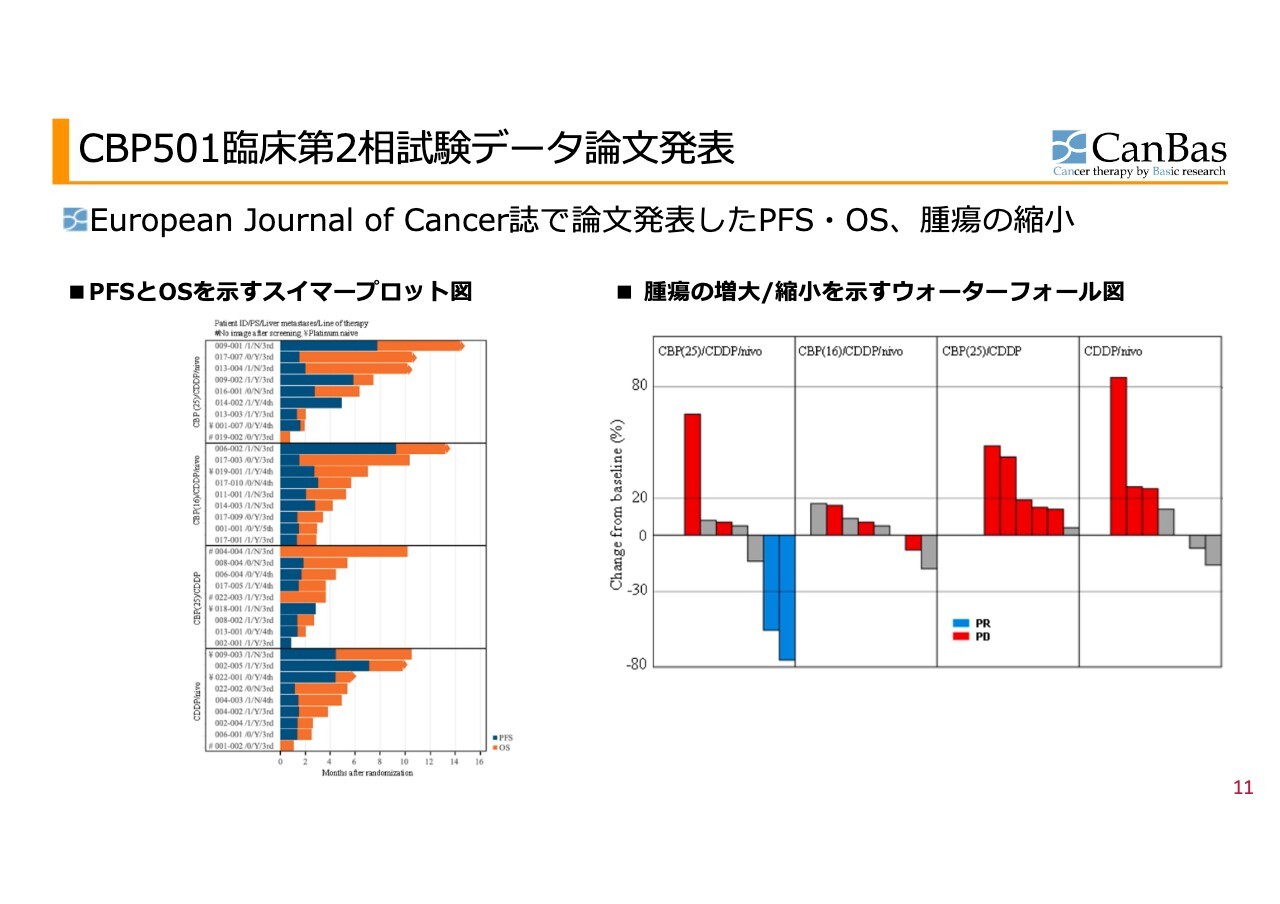
その結果のダイジェストです。スライド左のグラフは無増悪生存期間と全生存期間を示しており、それぞれの箱が試験における群を表しています。この試験は4群で行われました。
一番上が「CBP501」25mg、その下が「CBP501」の少量投与、下の2つは2剤併用群で、「CBP501」と「シスプラチン」の組み合わせ、および「シスプラチン」と「オプジーボ」の組み合わせです。
オレンジのバーをご覧いただくと、一番上が最も長いことがわかると思います。また、横棒の先端がとがっているのは、試験終了時点で患者さまがご存命であることを表しています。このため、この後どのくらい生存されたかについてのデータはありませんが、この時点ではご存命でした。
スライド下部を見ると、横棒の先端がとがっていない患者さまが大部分であることがわかると思います。
スライド右のグラフをご覧ください。このグラフは、治療を開始してから最もがんが小さくなった時点を示しています。バーが上向きの場合は、治療開始後にがんが大きくなったことを表し、バーが下向きの場合は、治療開始後にがんが小さくなったことを示します。
バーが下向きでマイナス30パーセントを下回る場合は「奏効」に分類されます。一方、プラス20パーセントを上回る場合は「PD(Progressive Disease/病勢進行)」と判断されます。
なお、がんの進行は必ずしも標的としているがんの大きさだけで決まるわけではありません。標的外の部位に新たながんが発生することで進行とみなされる場合もあります。そのため、がんの大きさが変わっていないにもかかわらず赤いバーが表示され、PDと判断される患者さまもいます。
4つの群の中では、グラフからも一目でわかるように、左側に示した最初の群において2名で奏効を示し、がんが非常に小さくなりました。奏効は膵臓がんの3次治療では非常に珍しく、一般には発生率が5パーセント未満とされているものです。
患者数は少ないですが、9名の患者さまの中で2名で奏効を示したというのは、一定のインパクトがある結果だと思います。さらに、実際に投与されていた患者さまは9名中8名であるため、現実にはそのうち2名のがんを小さくすることができたということになります。
2つ目の群では、青いバーで示された奏効には該当しないものの、全体的にがんが小さくなっている様子が見て取れます。
一方、残り2つの2剤併用群ではがんが大きくなっています。これらと比較して、先ほどの2つの群のほうが明らかに良い結果であることを感じていただけるのではないかと思います。
CBP501臨床第2相試験データ
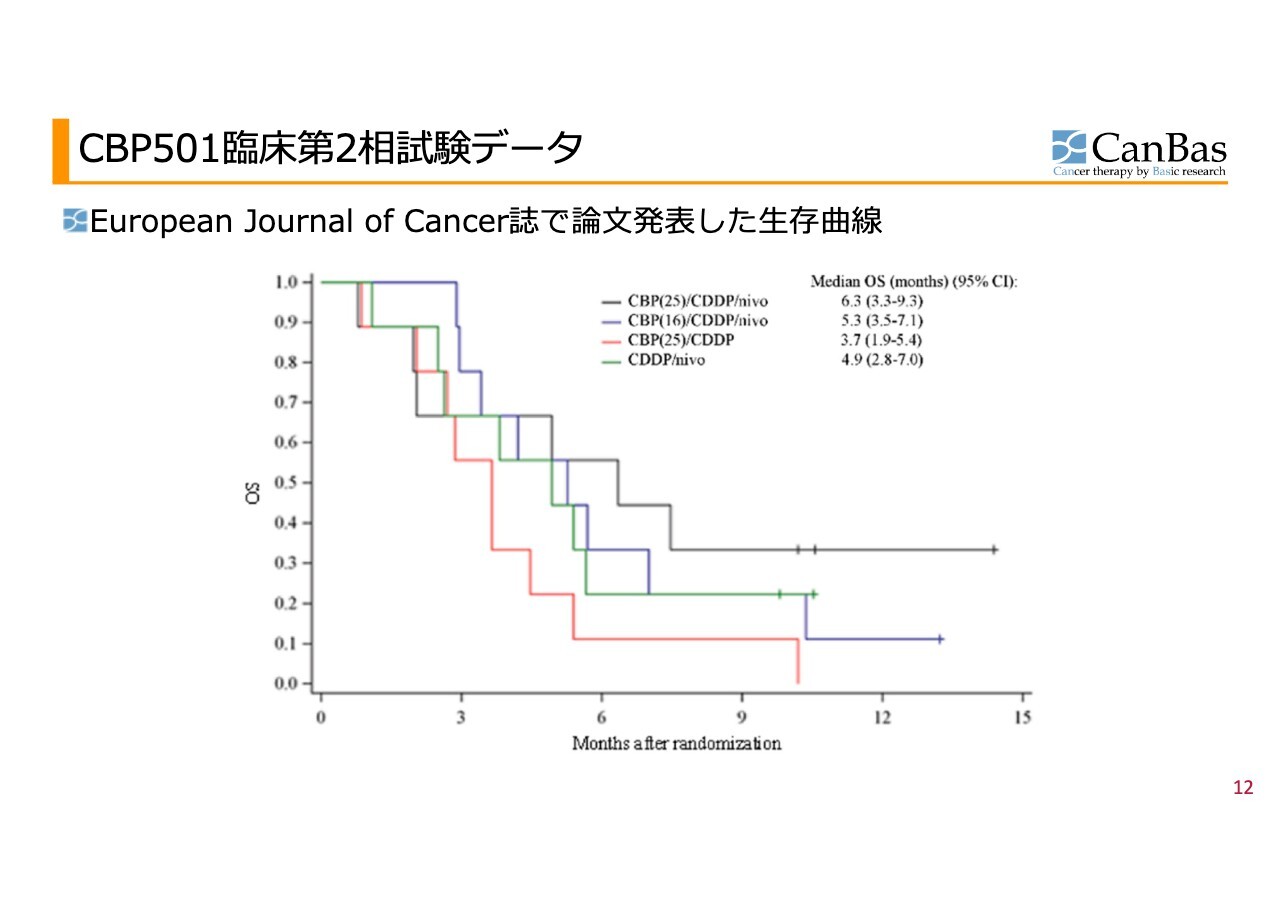
がんの試験で最も重要なのは全生存期間、すなわち患者さまの生存期間です。これを示すものとして、最初にご紹介したカプランマイヤー曲線があります。
カプランマイヤー曲線は、横軸が患者さまの生存期間、縦軸がその時点で生存している集団の割合を表しています。
この曲線を見ると、赤色の線で示されている「CBP501」と「シスプラチン」だけの治療が、この試験では最も悪い結果を示しています。しかし、3次治療における生存期間の中央値は3ヶ月といわれており、この赤色のカーブでも過去の臨床試験、いわゆるヒストリカルデータと比較すると、やや改善されている可能性があるといえます。
一方で、黒色の線で示される「CBP501」「シスプラチン」「オプジーボ」の3剤併用治療のカーブは明らかに上方に位置しており、特に6ヶ月以降の時点で生存している患者さまの割合が高くなっています。この結果は、当社が目指している理想的なカーブに近いものが得られたことを示しています。
目指してきた「最適な併用」への前進
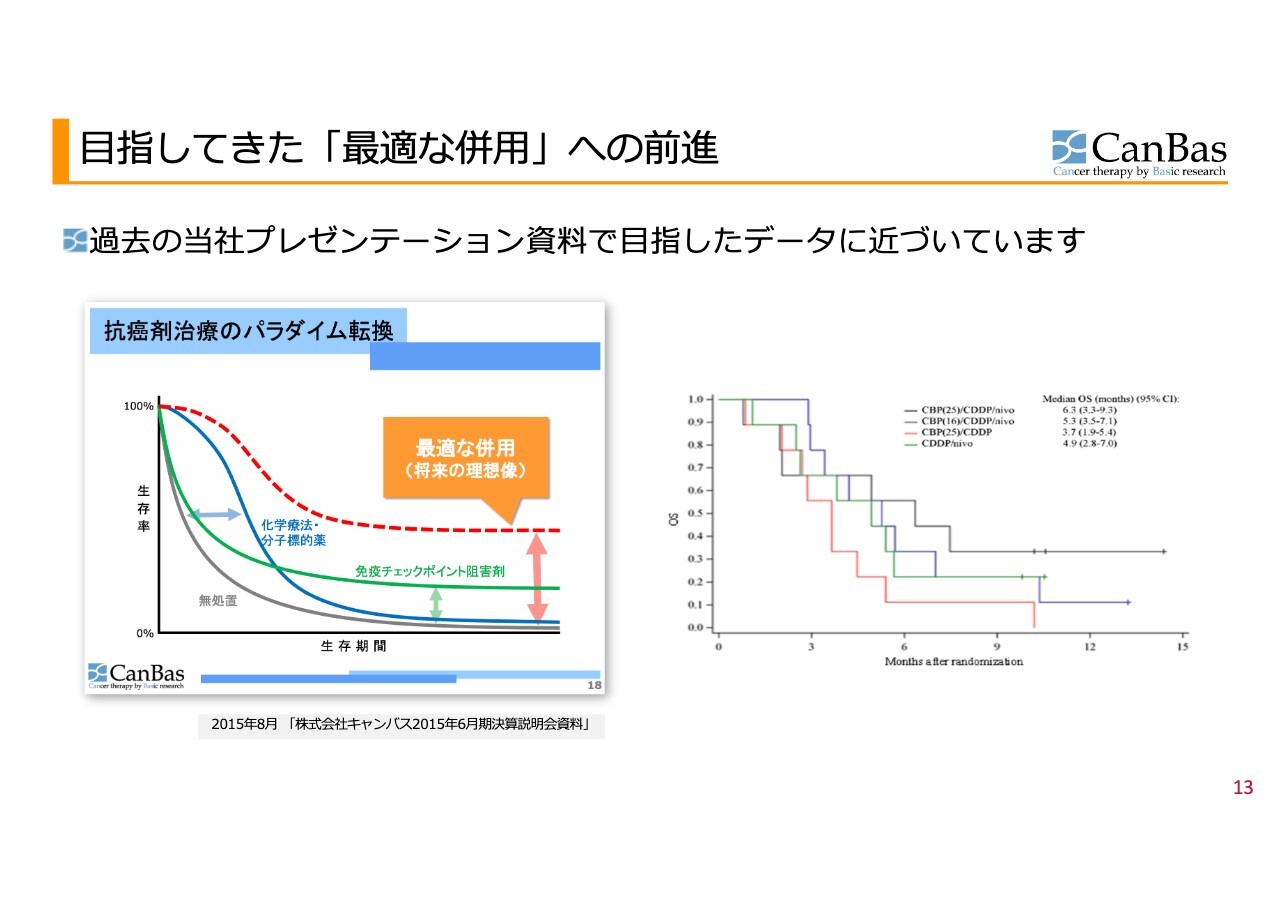
過去の当社プレゼンテーション資料で、将来目指すべきカプランマイヤー曲線として公表したのが、左のグラフです。これは2015年8月のもので、先ほどお話しした分子標的薬・化学療法を表す青い線と、免疫チェックポイント阻害剤を表す緑の線があります。
緑の線は残念ながら効果がある患者さまの数が少なく、「これを上に押し上げるのがこれからの目標です」とご説明したものが、この2015年当時の資料です。
右の実際の臨床試験データのカーブをご覧いただくと、まさにそのカーブのようなかたちが描かれていることがおわかりいただけるかと思います。
欧州第3相試験開始申請・準備の現状

この結果をもとに、最終的に「統計的にこれは間違いない」と示すための確認試験を実施したいと考えており、現在、欧州で臨床試験を行う準備を進めています。
欧州第3相試験開始の申請と準備の現状は、現時点でみなさまが最も関心を持たれている部分かと思います。欧州第3相試験とは、承認を得るための最終試験に該当し、この試験の実施に向けて現在申請手続きを行っています。
一方で、「開始承認取得の時期の不確実性」については、規制当局とのやり取りなど、当社ではコントロールできない時間的な不確実性が存在します。
次は「時期以外の不確実性」です。内容が不適切であったり、当局の意見と大きく異なる場合などで臨床試験を始められないような事態の発生リスクを「時期以外の不確実性」と呼んでいます。
現時点では、そのような不確実性が顕在化する恐れは低く、時期以外の不確実性として明確に懸念すべき事項はほぼないと考えています。
開始承認を受領してから開始に向かうための準備については、今回の最終試験は欧州で実施されることから多くの臨床試験施設と契約を結ぶ必要がありますが、これを着実に進めています。
「時期以外でも状況が悪化したときは米国移行を検討」と記載していますが、これは最終手段で、命綱のようなバックアッププランです。欧州で第3相試験がどうしても実施できない場合には、米国での第2b相試験に戻る準備があります。
しかしながら、現時点でそのような事態に陥る兆しはなく、その必要はないと考えていますので、欧州での臨床試験開始を最優先とする方針で、現在活動を進めています。
第3相試験の費用見通しは、現時点で45億円から50億円としています。初期の目標の実現可能性については、現時点で変化はありません。
引き続き経営資源を欧州での臨床試験開始に集中しており、米国については、臨床試験の第2b相をいつでも開始できる許可を得ている状況のまま、進行を保留しています。
時期以外の不確実性への対応
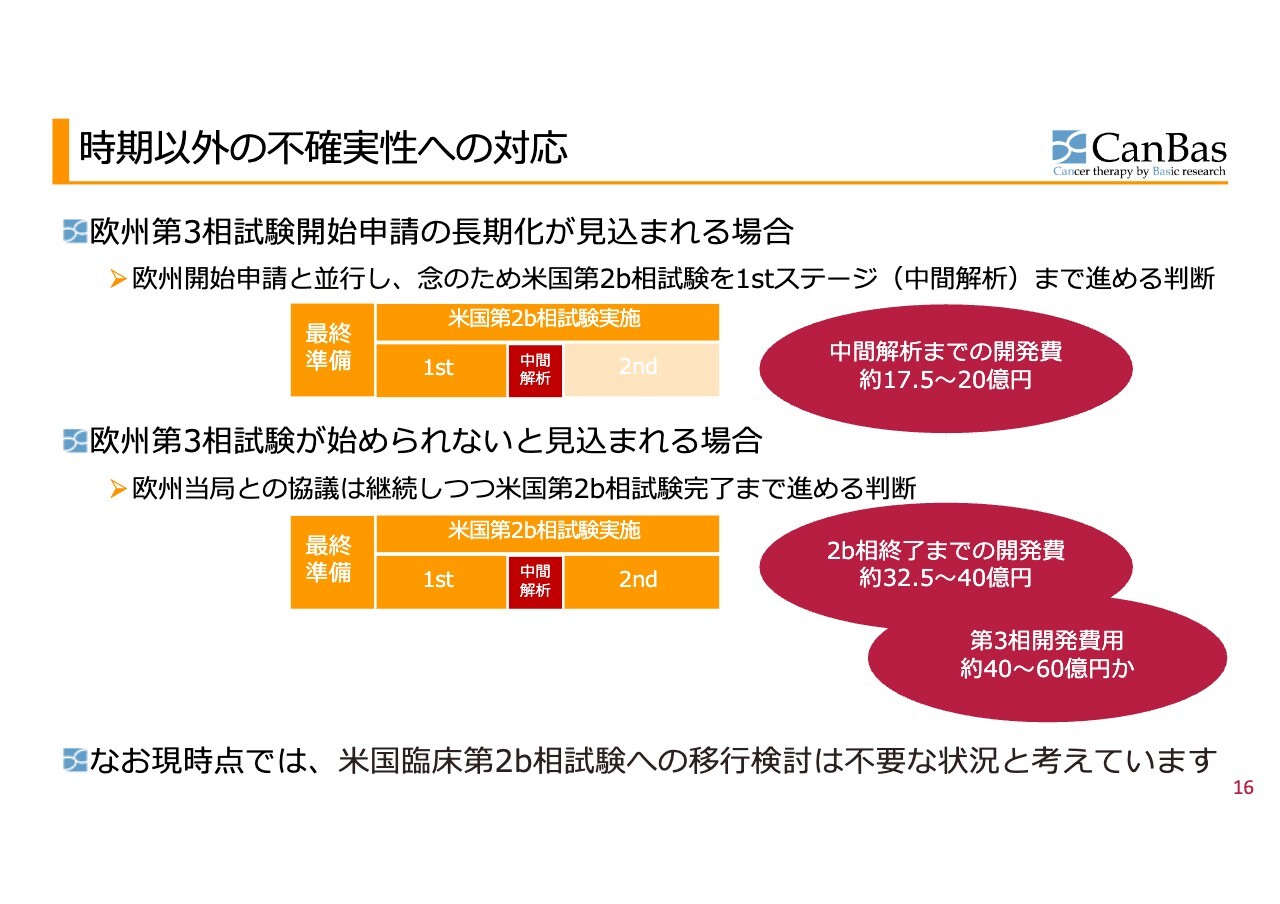
欧州第3相試験開始申請が長期化する可能性がある場合の、不確実性への対応についてご説明します。
もし許可が下りるまでに時間がかかると見込まれる場合に、欧州試験と並行して米国の第2b相のファーストステージのみを実施した場合の費用をスライド上部に示しています。
一方、欧州第3相試験が開始できない場合については、スライド下部に示しています。欧州を断念して米国で第2b相試験を終了させ、その後第3相試験まで実施する場合、このような多額の費用が必要になる見込みです。
しかし、現時点では米国第2b相試験への移行を検討する必要はないと考えています。欧州で第3相試験を実施できる見込みがあるので、現在も欧州への注力を継続しています。
XPO1阻害による抗がん活性のしくみ
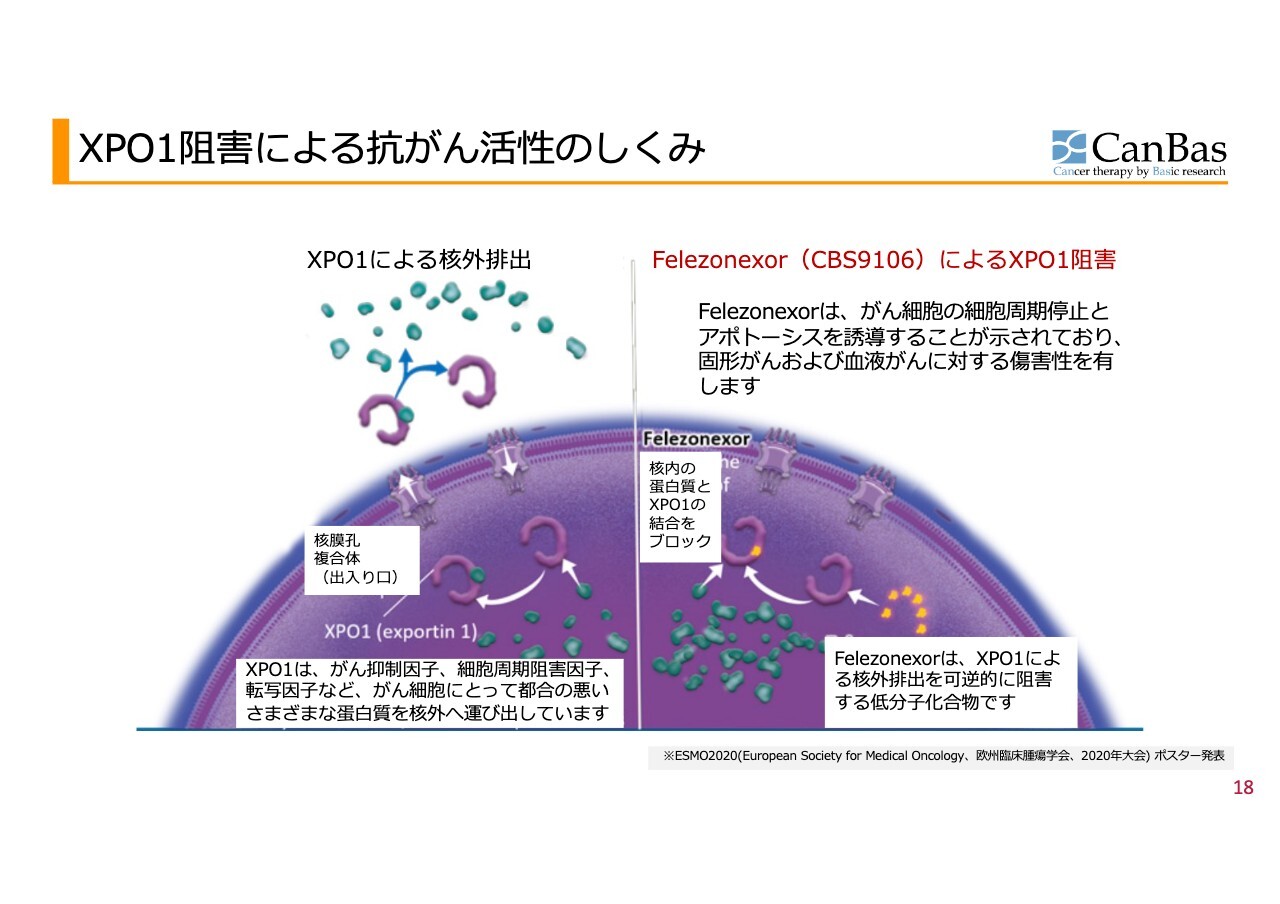
次のパイプラインである、可逆的XPO1阻害剤「CBS9106」について少しお話しします。
細胞の中には核という部分があります。この核から細胞質へタンパクがコントロールされた状態で運ばれる際、XPO1(exportin1)という特別なタンパク質が関与しています。XPO1は核から細胞質へ、タンパク質やメッセンジャーRNAなどを輸送します。
がん細胞にとって不利ながん抑制タンパクの多くは核内で機能するため、がん細胞はこの仕組みを利用して核内からがん抑制因子を排出し、生存しようとする場合が比較的多く見られます。
そこで、このXPO1を一時的に停止させることで、がん抑制因子ががん細胞を死滅させるというのが、XPO1阻害による抗がん活性の仕組みとなります。
CBS9106 ライセンス契約解消・全権利返還と今後

「CBS9106」については、米国のStemline社とライセンス契約を結び、2014年12月に日中台韓を除く全世界を対象にライセンスを導出しました。その後、2018年には対象地域を拡大し、前述の4ヶ国を含めました。
その後、同社は臨床試験第1相を終了し、第2相の準備を進めていましたが、2025年6月にライセンス契約を解消し、すべての権利を当社に取り戻すこととなりました。
ライセンス契約に基づき受領した収益としては、契約一時金および技術アドバイザリーフィーがあり、アドバイザリーフィーについては累計7億円程度を受領しました。
今後の展開についてですが、抗がん剤に関する世の中の状況が日々めまぐるしく変化する中で、本分子の作用や薬剤の特徴は非常に明確です。そのため、どの領域でどの試験を実施すべきかを見極め、確実に目標を定めて進めていきたいと考えています。
また、追加で行う基礎研究の成果や当社の財務状況などを総合的に考慮し、開発方針を固めていく予定です。
CBS9106の優位性

「CBS9106」の優位性についてですが、「CBS9106」と同じ作用機序を持つ薬剤XPOVIO(一般名Selinexor)が、米国のKaryopharm社からすでに上市されています。この薬剤は、びまん性大細胞B細胞性リンパ腫や多発性骨髄腫といった血液系のがんに対して承認を得ています。
ただし、副作用が比較的強いため、その回避に向けた工夫が重ねられ、最近ではかなり改善されています。
一方、「CBS9106」については、前臨床試験や基礎研究データから、XPOVIOが持つ副作用に比べて、副作用が少ない可能性があると当社は考えています。
また、「CBS9106」については、XPO1分子を分解に導くことで、副作用の小ささにつながっていることを発見しています。そのため、Karyopharm社のXPOVIOと比較しても、高い優位性があると考えています。
CBT005の構造
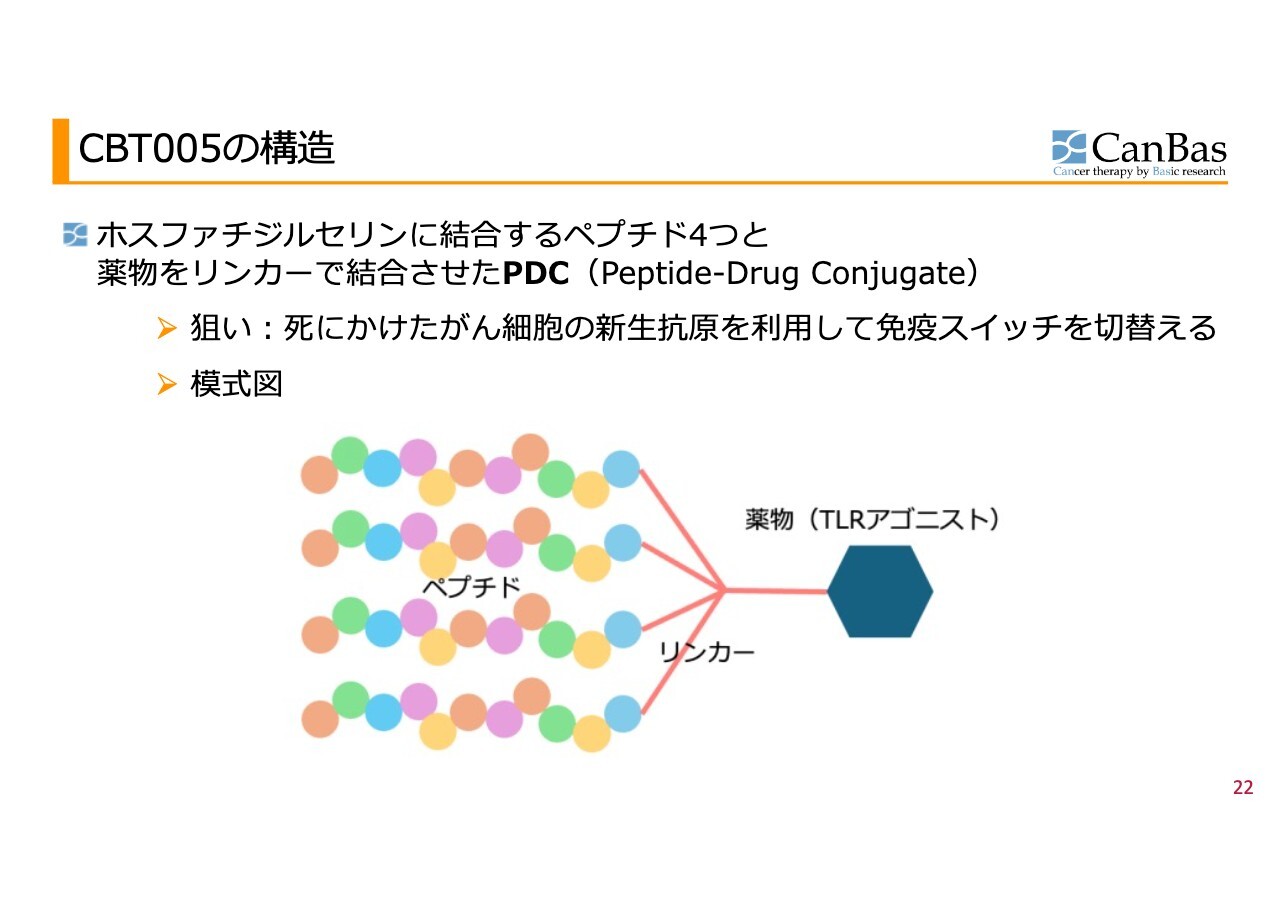
3つ目のパイプライン「CBT005」は、前臨床に向けて動き出した化合物のパイプラインです。
これはまさに免疫系を利用するもので、どちらかといえばワクチンに近い側面があります。死にかけたがん細胞に結合し、そのがん細胞を免疫細胞が取り込んだ後、それを免疫細胞に攻撃するものだと示すように導く薬剤です。この仕組みを「ImmunoSwitch」「免疫スイッチ」と呼んでいます。
CBT005の作用機序
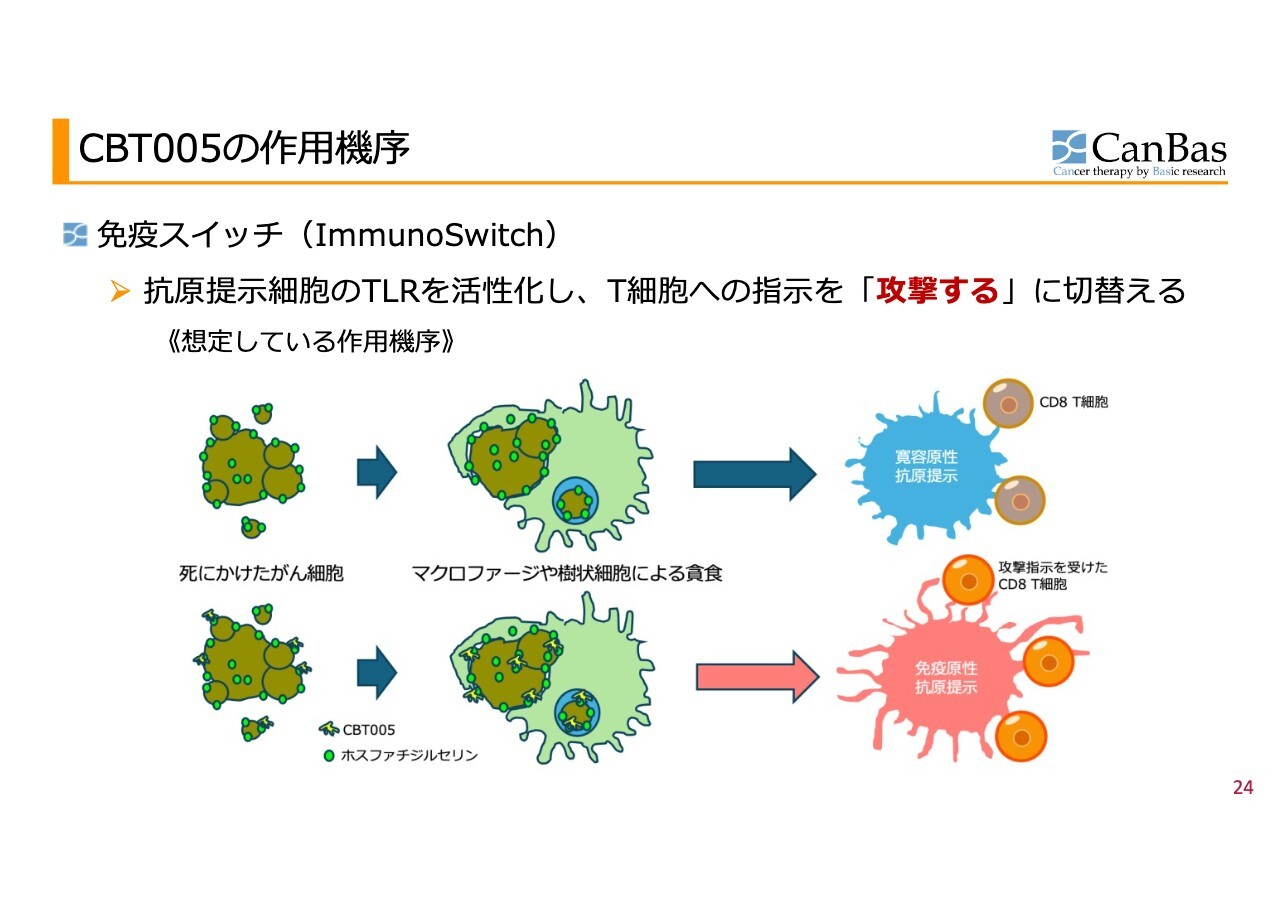
抗原提示細胞というのは死にかけたがん細胞を食べる細胞です。死にかけたがん細胞をマクロファージや樹状細胞が食べ、その後T細胞に情報を提示します。
スライドの上部に「免疫寛容性抗原提示」と記載しています。がん細胞の特徴として「攻撃しなくていいよ」や「攻撃しちゃいけないよ」という提示を行う点があります。
これが、がん細胞が「治らない傷」といわれる理由の1つです。がん細胞を取り込んだ免疫細胞が「もうここでの攻撃はやめましょう」と言ってしまうのです。
マクロファージや樹状細胞ががん細胞を取り込む際には、ホスファチジルセリンを目印として利用します。当社の「CBT005」には、このホスファチジルセリンに結合する特性を付与しています。
がん細胞を取り込んだ後に、「CBT005」に結合している自然免疫を活性化する物質が同時に取り込まれることで、スライド上の免疫寛容性抗原提示から免疫原性抗原提示へと変わり、「こいつを攻撃しろ」と命令を出すようになります。
この仕組みをワクチンに例えると、がん細胞が持つ特徴の一部を人に投与するのではなく、がん細胞そのもの全体をワクチンのように利用できるという見方も可能となります。
このような作用を持つ物質です。つまり、これもCBP501と同様に「免疫コールドながん」を「免疫ホットながん」に変える薬剤ということになります。
基礎研究〜非臨床試験段階のプロジェクトの状況

臨床試験前段階の研究開発についてご説明します。前臨床試験前の段階ではパイプラインとは呼ばず、研究段階とされます。
すでにご紹介したもの以外にも、「IDO/TDO二重阻害剤」という、がんの局所で重要な免疫抑制作用を解除する可能性のある薬剤があります。
また、「CBP-A08」は「CBP501」の後継となる薬剤です。
「NEXTプロジェクト」は免疫系細胞の特異的な活性化を助ける有力なメカニズムを持っています。メカニズムとしては有力なのですが、まだ研究段階にあります。
さらに、まったく異なる種類の研究として、「抗がん剤感受性予測システム」というコンピューターを使用した感受性予測システムの研究も進めています。
以上、これまでの開発の経緯と現状についてご説明しました。
2025年6月期の業績 (1)損益計算書
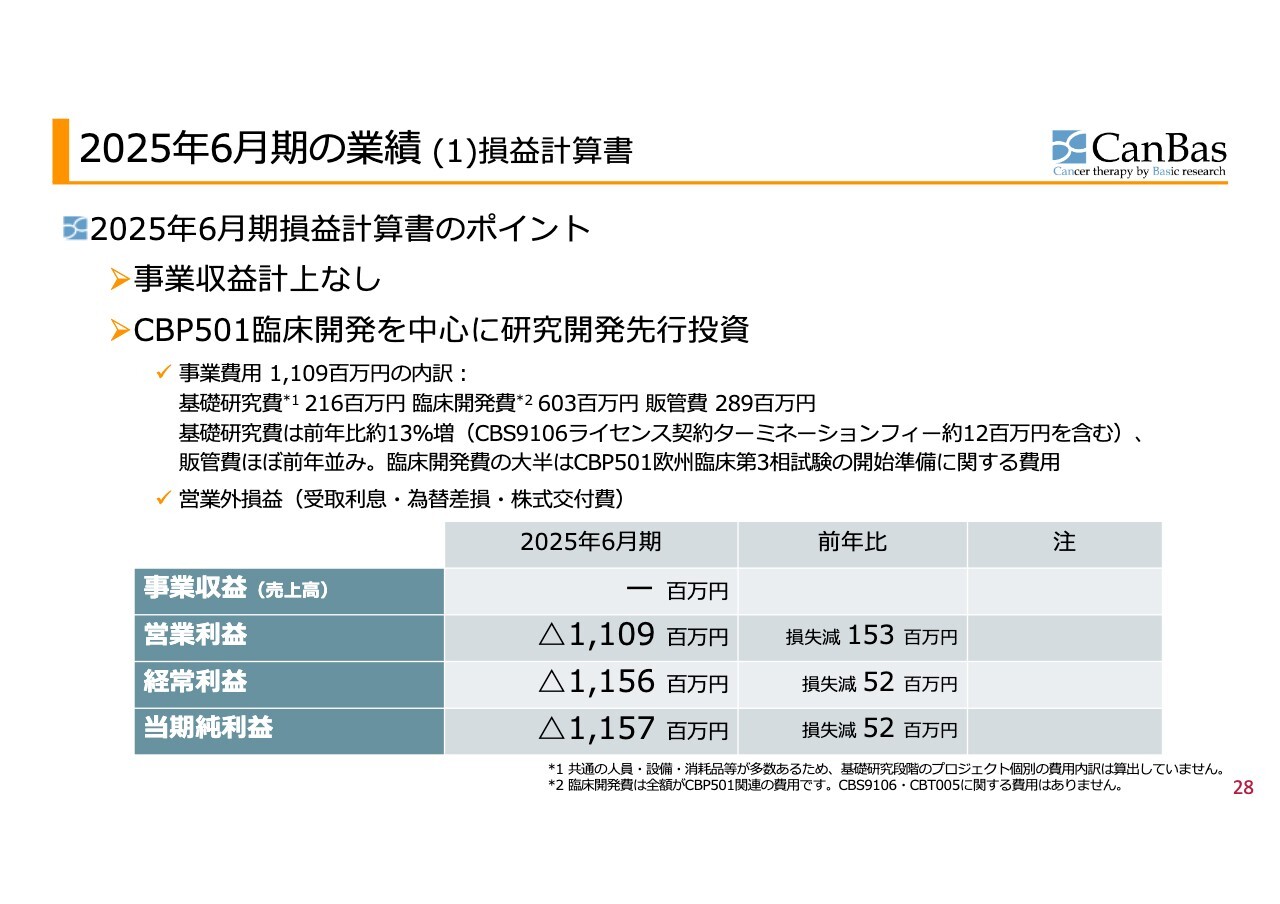
加登住:私からは2025年6月期の決算および財務状況についてご説明します。
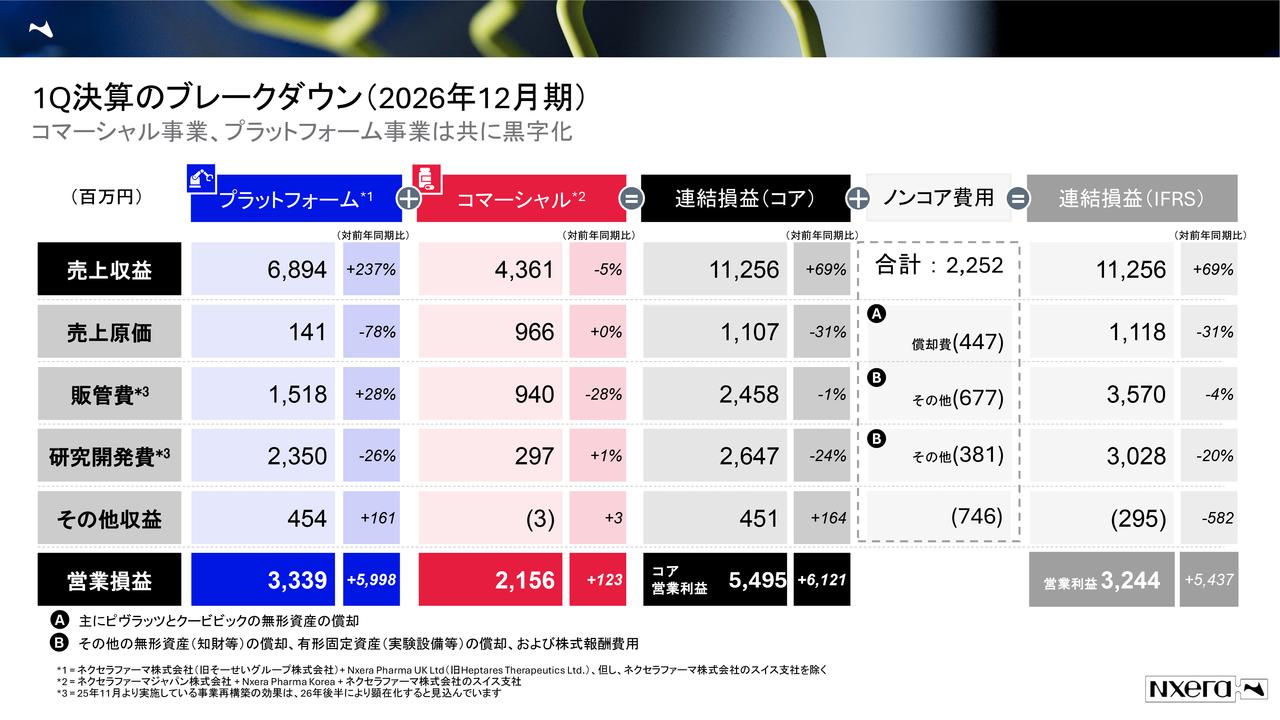
いつもと同様に、損益計算書のポイントについて申し上げます。事業収益の計上はありませんでした。事業費用は11億900万円で、内訳は基礎研究費が2億1,600万円、臨床開発費が6億300万円、販管費が2億8,900万円です。
基礎研究費については前年比で約13パーセント増加していますが、今年6月末に「CBS9106」のライセンス契約を終了した際のターミネーションフィーとして、Stemline社に支払った約1,200万円を含んでいます。これを考慮すると、大きな増加ではなく一時的な要因と言えます。
販管費2億8,900万円は、ほぼ前年並みの水準です。
臨床開発費については、その大半が「CBP501」臨床第3相試験を欧州で推進するための費用です。特に、薬剤の製造、製剤化、供給などを規制に適合させるための準備等に関連するコストが中心となっています。
その他営業外損益については、同時に公表したとおりです。
事業費用は主に臨床試験費用で増減
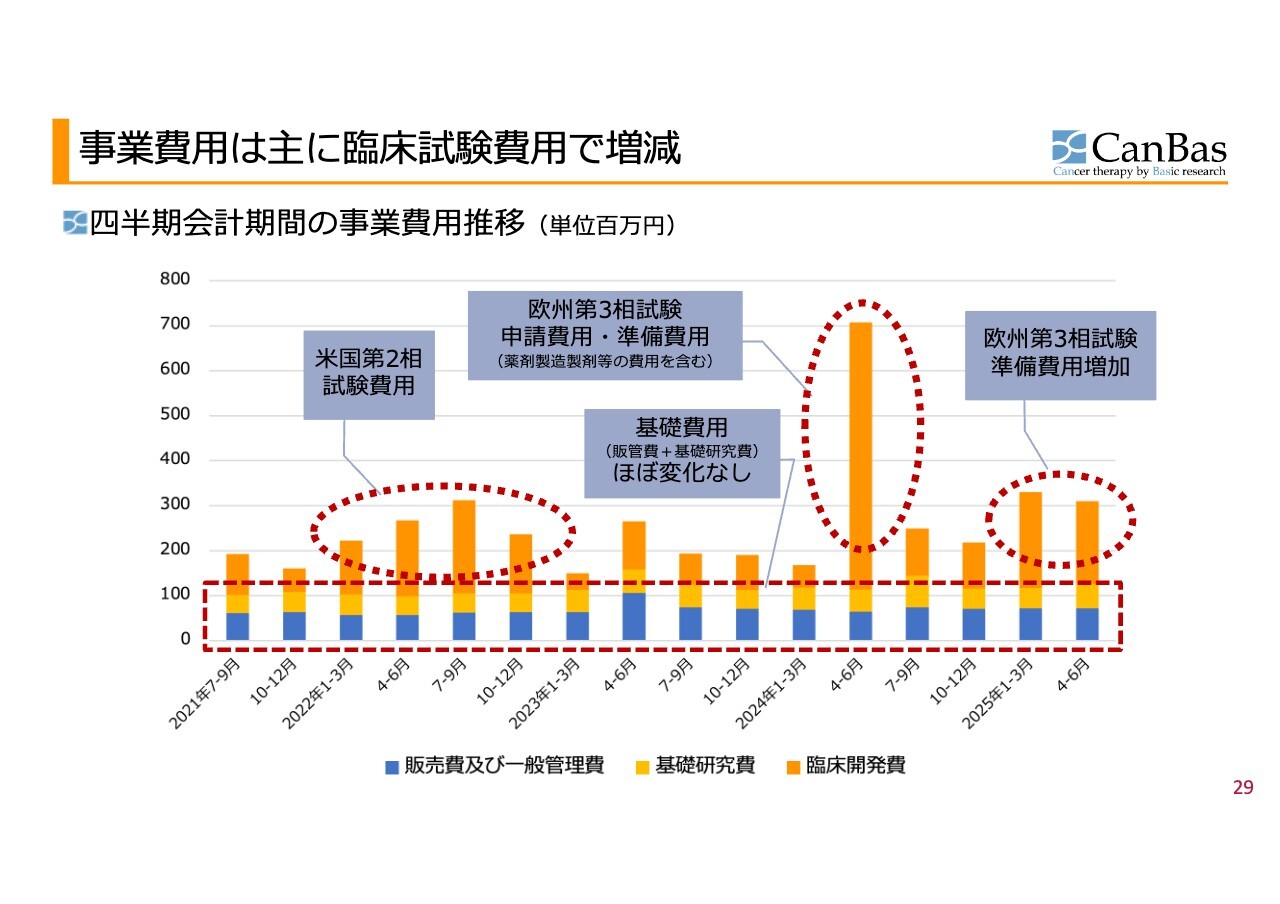
四半期会計期間の事業費⽤推移です。四半期ごとのデータをまとめることで非常にわかりやすくなると考え、毎回ご報告しています。
過去4年間の四半期の推移をご説明していますが、2024年4月から6月の棒グラフで目立っている部分を除くと、スライド内の棒グラフにおける青い部分の販管費と黄色い部分の基礎研究費は、四半期あたり1億円から1億2,000万円程度で、毎年ほぼ安定的に推移しています。すなわち、年間では約4億円から5億円がキャンバスにおける基礎的な支出のベースとなっています。
それに加え、棒グラフのオレンジ色で示した臨床開発費はその時々に抱える臨床開発プロジェクトに応じて変動する出費として計上されています。2022年半ば頃、米国で第2相試験を実施していた際には支出が一時的に増加し、グラフ上では山ができています。
また、棒グラフが突出している2024年4月から6月の四半期に関しては、第3相試験の申請が進行していた時期であり、申請自体の費用や準備費用に加え、薬剤の製造や製剤の取り組みが進められたことにより、この四半期で大きな支出が発生しています。
なお、これが臨床試験全体のピークというわけではなく、臨床試験開始時のイニシャルコストが集中した結果といえます。
直近の準備費用については、引き続きブログなどでもご紹介していますが、製造・製剤化に関連する規制適合や規制対応、さらに規制当局とのやり取りへの回答といった部分でコストがそれなりにかかっています。ただし、特段異常な値ではないと見ています。
2025年6月期の業績 (2)貸借対照表の推移

ご注目いただきたいバランスシートについてです。
2024年6月期末との比較で現預金を約9億6,000万円増加させることができました。当期純損失による出費はありましたが、2023年に実施したファイナンスの進展により、キャッシュが増強されています。
現在の企業価値評価に対するキャンバスの認識
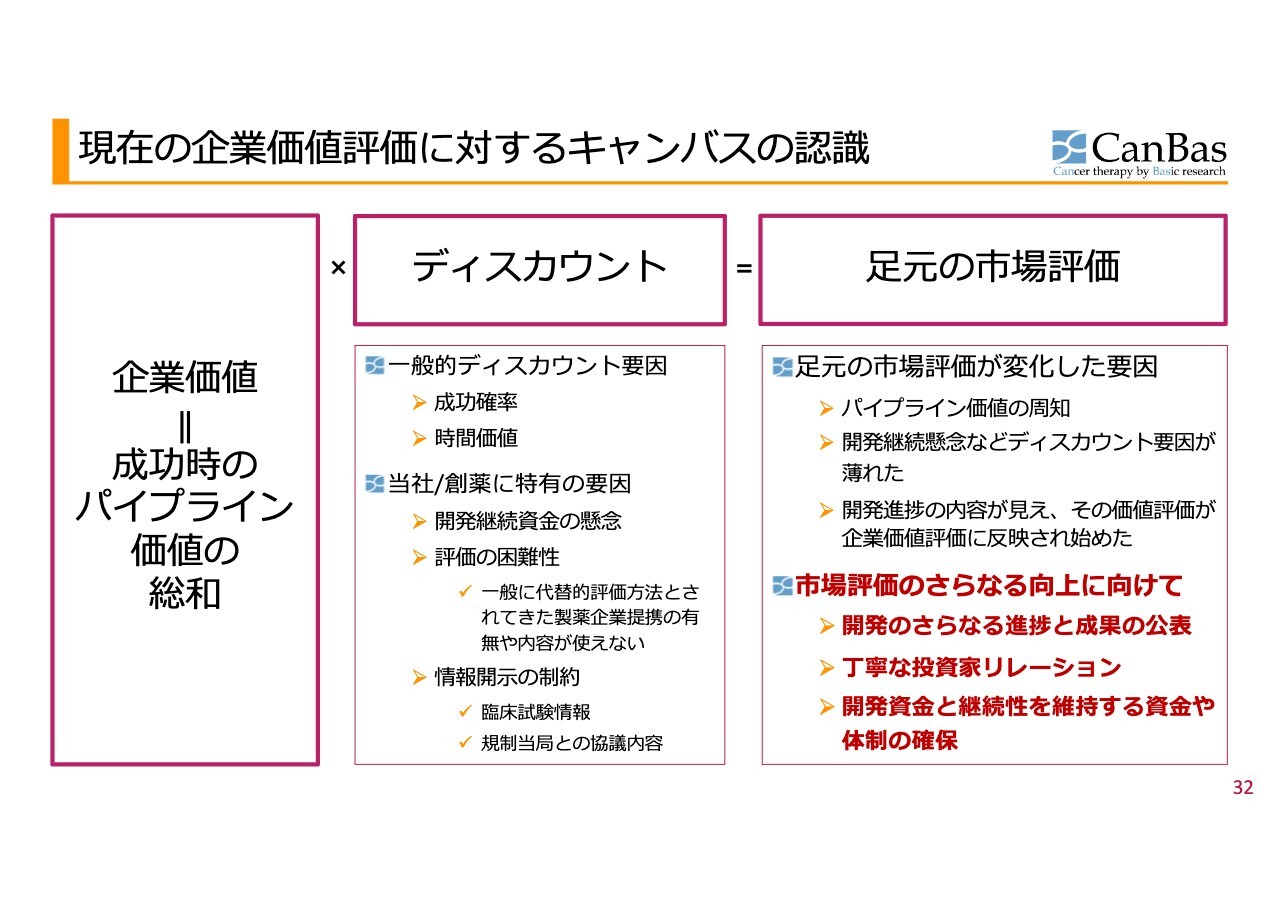
企業価値評価についてです。2022年頃からスライドに示されている表を用いて説明してきました。現在、株式市場において当社の企業価値は、時価総額約200億円と評価されています。
当社のような企業の企業価値は、おおむね成功した際のパイプライン価値の総和で考えられるべきですが、これに対してディスカウントがかかるのは当然と言えます。
Net Present Value(NPV)の一般的なディスカウント以上に、当社や創薬という領域特有の要因がいくつも存在しており、その結果として現在の市場価値が形成されていると考えています。
現状の時価総額約200億円というご評価は、臨床第3相試験の待機期間中である状況も考慮すると、十分にご評価いただけているのかは、まだつかめていない部分もあります。当社をご理解いただいた上で評価していただくために、開発状況や進捗をしっかりとご報告していきたいと考えています。
また、現状のように規制当局との対応に時間がかかり、開発が停滞している状況においては、丁寧な投資家リレーションを進めることでご理解をいただきたいと思います。
開発資金、継続性を維持するための資金、開発体制確保、この3つがポイントであると考えています。
CBP501パイプライン現在価値評価を動かすもの
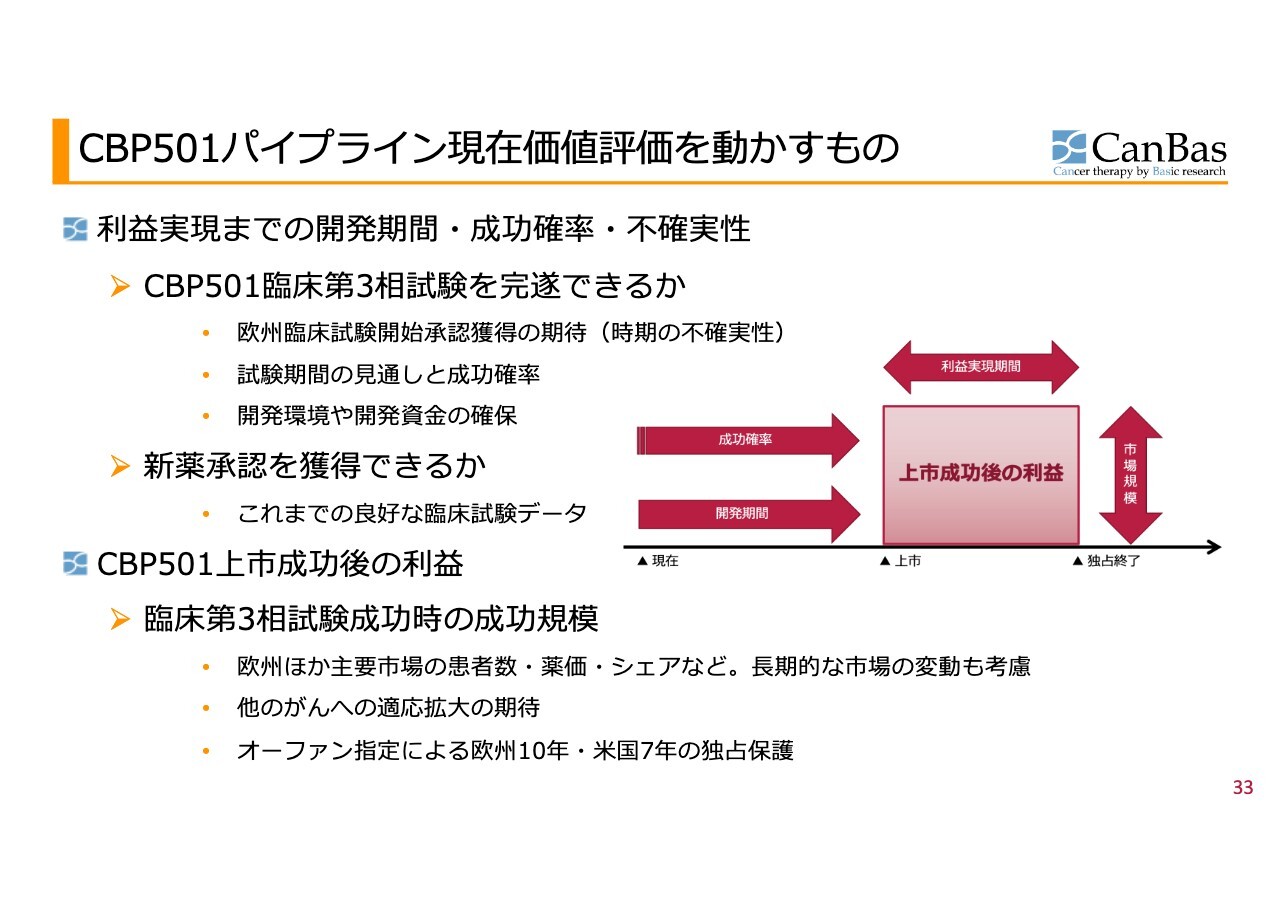
この3つのポイントのうち、開発資金の獲得については、第3相試験を開始するために必要な資金が確保できているというのが、先ほど河邊の話にあった現状の認識です。
パイプラインの現在価値評価は、スライドに示した内容に基づいて動いていると考えています。これらの4つの矢印を意識し現在価値を大きくするために、日々活動を続けています。この図については、日常的な投資家リレーション中でも触れているため、ここで詳細な説明は省略します。
開発資金確保に関する当社の現状認識

2023年に実施した開発資金の確保に関してあらためて申し上げると、ファイナンスはおおむね成功裏に完了しました。第三者割当増資と新株予約権の行使により、約40億円を調達しています。
当初の目論見としてはより大きな額の調達を目指しており、その額には及ばないファイナンスとなりましたが、最低限の目標である臨床第3相試験の開始資金の確保については問題なく進めることができました。結果として、現状28億円を超えるキャッシュを保持して決算期を越えることができています。
第3相試験の資金需要について、当初の見込みは45億円から50億円となっています。現状、プロトコル等に関して規制当局との間で大きなディスカッションは発生しておらず、その見通しに基づいて、当初の見込み費用である45億円から50億円に現時点で変更はありません。このうち一部はすでに支出済みであり、今後の出費は想定より小さくなる可能性もあります。
この見込み費用の全額を現時点でカバーするだけのキャッシュは持っていないのは事実です。そのため、状況を見ながら追加で資本政策を検討する必要があります。
資本政策としては、市場からの資金調達、あるいはなんらかの提携等による製薬企業などからの資金調達、または市場を直接介さない、将来的な提携を見越した第三者割当増資による調達などが考えられます。
ただし、現在のような第3相試験の開始が不確実である現状下において、時価総額が比較的低めで評価されている現在のタイミングでは、資金調達を急ぐべきではないとも考えています。
そのため、現状においては開始承認に関する具体的な動きがないままで資金調達を慌てて進める状況ではないと判断しています。今後追加で資本政策を実施する際は、株主価値の毀損や希薄化を可能な限り回避したいと考えています。
今後想定されるCBP501関連ニュースフロー

今後想定されるニュースフローについて、変更はありません。ただし、時期については当社ではコントロールしきれない不確実性があることをあらためてご理解いただきたいと思います。
キャンバスを知る情報源

こちらの情報源についても変更はありません。以上で決算説明会の説明を終了します。
質疑応答:臨床試験の開始および進行における不確実性と2027年目標について
加登住:「2027年の上市承認目標を変えていないのはどのような背景なのでしょうか? あるいはどのような時期になれば修正するのでしょうか
新着ログ



「医薬品」のログ