2026年3月期決算説明
Delta-Fly Pharma、「DFP-10917」単剤で米国FDAと条件付き承認協議へ TP53含む予後不良遺伝子変異患者で有効性示唆
2026年3月期のサマリー
江島淸氏(以下、江島):Delta-Fly Pharma株式会社代表取締役社長の江島です。2026年3月期のサマリーの中から、研究開発の取り組み方とパイプラインの進捗についてご説明します。
研究開発の取り組みについては、開発パイプラインが6本あり、そのうち5本が臨床試験を進めている段階です。各パイプラインにおける特許承認の取得や申請、更新といった選択と集中の取り組みについても簡単にご説明します。
パイプラインの進捗については、臨床第3相試験(臨床第2相試験/臨床第3相試験含む)と臨床第2相試験、この2つの段階において臨床試験を進めています。
臨床第3相試験の取り組みとしては、「DFP-10917単剤」「DFP-14323」「DFP-17729」があります。
臨床第2相試験については臨床第1相試験/臨床第2相試験も含めて取り組んでおり、その中で「DFP-10917とベネトクラクス(VEN)の併用」、「DFP-14927」はアメリカで、「DFP-11207」は日本で実施しています。
開発パイプラインの状況

詳細についてご説明します。
「DFP-10917」という点滴静注剤について、再発・難治性の急性骨髄白血病患者を対象に実施した治験です。臨床第3相試験の中間解析が終了しました。この中間解析の結果を受け、安全性独立委員会であるDSMBの提言に従い、試験そのものをいったん停止しました。
試験を停止しなければデータの解析ができず、対照群との差異を確認できないため、試験を止めた次第です。なお、安全性に関する問題があったわけではありません。
その結果、私どももようやくデータの詳細を確認できるようになりました。そのデータを入念に精査したところ、有意差は確認されませんでしたが、奏効率(効果の指標)は対照群よりも良好でした。
対照群には強化療法と非強化療法の2群がありますが、強化療法(一般的な療法)群との層別解析において、全生存期間は「DFP-10917」群の方がより高い結果となりました。
特にDSMBの先生方から強く推奨された項目ですが、「DFP-10917」の単剤は、TP53を含む予後不良の遺伝子変異を有する患者集団において有効性を示唆する結果でした。
多くの患者で「DFP-10917」が効果を示したことから、この3つの項目を踏まえ、有意差がなかったとしても非常に価値のある臨床データとして、現在FDAに条件付きの申請を進めています。
「ベネトクラクス(VEN)」併用試験に関しては、臨床第1相試験/臨床第2相試験がすべて終了し、第2相試験部分の症例登録も先頃完了しました。フォローアップは現在も継続中です。
データモニタリング委員会の結果によれば、安全性についてはまったく問題がなく、認容性も高いとの判断をいただいています。
臨床第1相試験での奏効率は50パーセントであり、CR1例とMLFSの評価項目も含め、安全性と効果の両面で良好な結果が得られました。
効果を評価する臨床第2相試験では、奏効率が53パーセント、CR相当のCRiが3例、MLFSが3例、さらにPRが3例という結果が得られています。
これらのPR3例も、基本的には「ベネトクラクス(VEN)」の併用レジメンが1回施行された後の患者に確認されたものであり、このPRにも非常に意味があるとのコメントを臨床の先生方からいただいています。
この状況を受けて、FDAとこの結果をもとに次の臨床第3相試験に入るのではないかと考えています。そのため、FDAとの臨床第2相試験終了時相談(End of Phase 2 Meeting)の開催を近々に計画しています。現在、さまざまな資料作成や準備を進めています。
日本においては、以上の状況を踏まえたうえで、日本新薬が臨床第1相試験を現在も継続中です。
開発パイプラインの状況

「DFP-14323」は経口剤で、対象は末期の肺がん患者です。日本では臨床第3相試験に入っており、現在は順調に進んでいます。国内では、臨床30施設で症例登録が鋭意継続中です。
「DFP-17729」も経口剤で、末期の膵臓がん患者を対象に臨床第2相試験および臨床第3相試験を実施しています。現在は、臨床第2相試験/臨床第3相試験のうち、第2相試験部分の症例登録を進めています。
この試験は、日本で前例のないアダプティブ・シームレスデザインを採用しており、エンドポイントである生存期間を効率的に検証できる仕組みです。
また、臨床第2相試験/臨床第3相試験の中で確認された優越性の程度に基づき、臨床第3相試験の症例数を調整します。臨床第2相試験で効果がかなり良好であれば、臨床第3相試験で必要な症例数が減少する設計となっています。本年7月頃には症例登録がすべて完了する見込みです。
データ解析は症例登録が完了した後、数ヶ月間で行われ、その後、臨床第3相試験に移ります。このプロセスでは、症例数の増減などを含むさまざまな準備を進める必要がありますが、数ヶ月程度で速やかに次の臨床第3相試験に移行できる見通しです。
「DFP-11207」は、胆道がんを対象とした経口剤であり、医師主導治験として実施されています。当社自らが治験を実施しているわけではなく、臨床の先生方にお願いして進めている試験です。
この試験については、一般社団法人日本肝胆膵オンコロジーネットワークとの共同プロジェクトとして、当社が臨床治験薬の提供とサポートを行いながら、臨床第1相試験/臨床第2相試験を進めています。
臨床施設としては、神奈川県立がんセンター、大阪国際がんセンターに加え、埼玉県立がんセンターで実施する予定です。
「DFP-14927」は、「DFP-10917」のDDSであるポリエチレングリコールの誘導体であり、米国で臨床第1相試験の拡大試験を実施中です。この拡大試験は、当初大腸がんを対象としていましたが、より迅速かつ効果的に進めるため、現在は膵臓がん患者を対象としています。
GEM/nab-PTX併用療法が無効の膵臓がん患者を対象に、病勢コントロール率(DCR)の改善が25パーセント以上となれば、次の第2相試験、または臨床第2相試験/臨床第3相試験に進む見通しです。
以上で、私からのご説明は終わります。
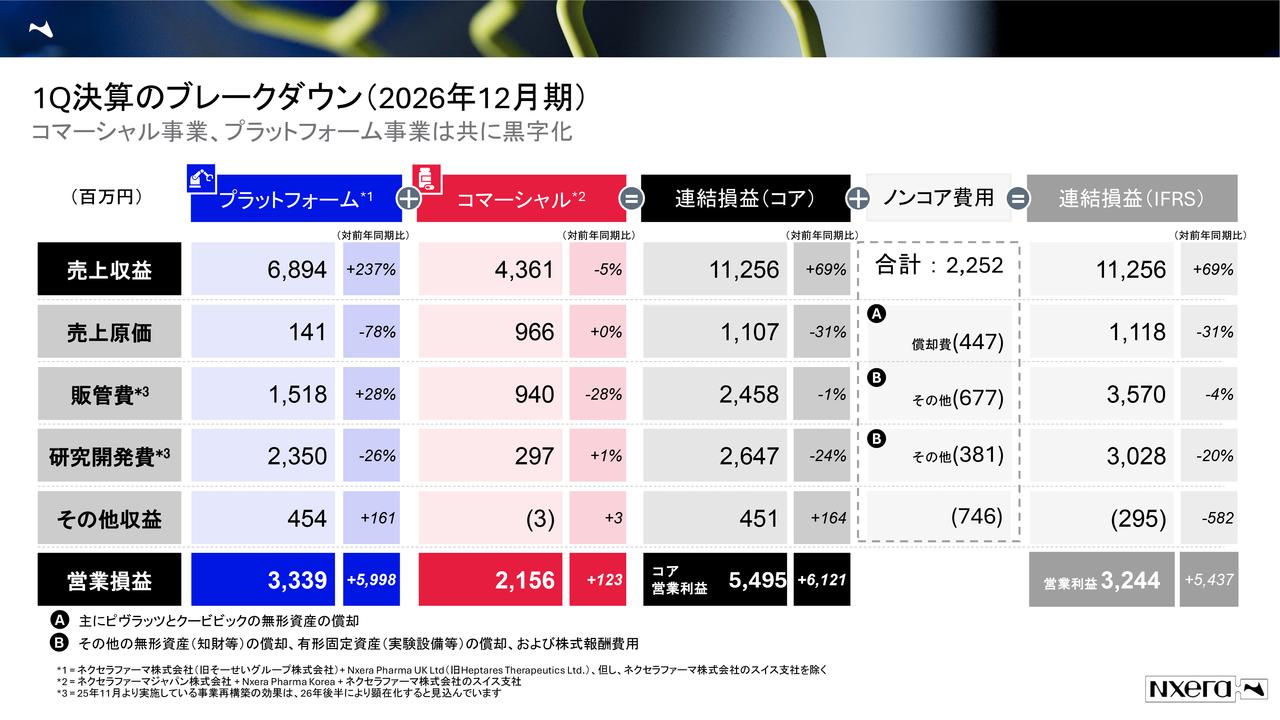
2026年3月期 通期決算の概略
黒滝健一氏(以下、黒滝):取締役管理部門担当の黒滝です。2026年3月期の決算概要と、2027年3月期の予想についてご説明します。
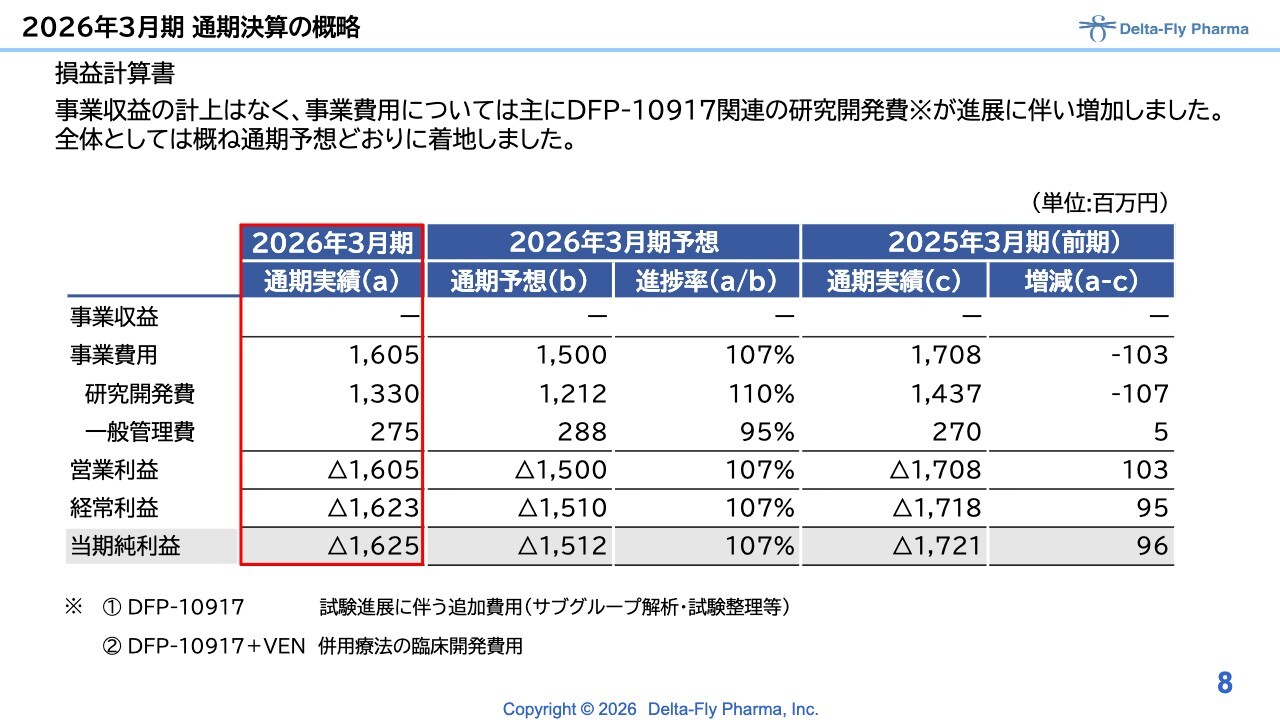
2026年3月期について、損益計算書において事業収益はありませんでした。先ほど江島から説明があったとおり、当社のパイプラインの開発を進めたことが主な内容です。特に事業費用に関しては、「DFP-10917」関連の研究開発が進展した影響もあり、増加しました。
全体としては概ね通期の計画どおりに着地しましたが、事業費用は計画に対して107パーセントかかりました。
内訳として、研究開発費が計画比110パーセント、一般管理費が計画比95パーセントとなっています。営業損失、経常損失、当期純損失はそれぞれ16億円を超えており、計画比でいずれも107パーセントを上回る結果となりました。
2026年3月期 通期決算の概略
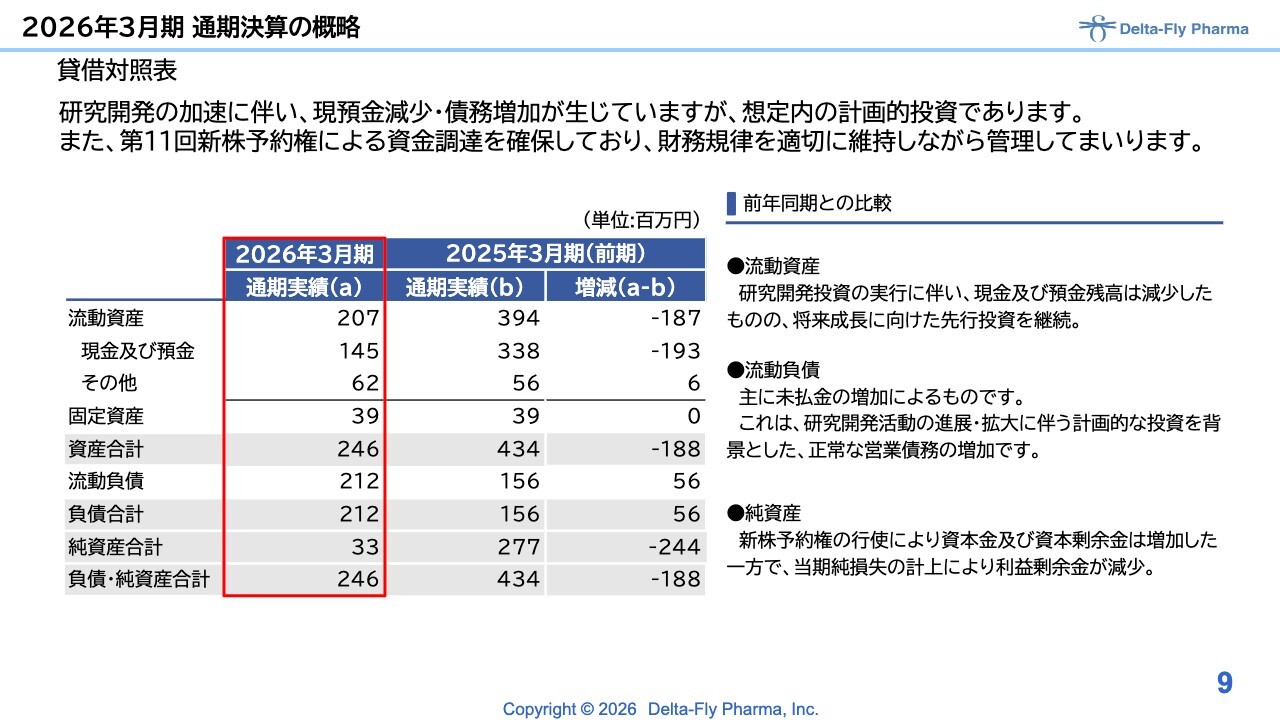
貸借対照表についてご説明します。
研究開発の加速に伴い、現預金の減少と債務の増加が生じていますが、これらは計画の中で想定内のものであり、投資を進めてきました。一方で、現預金の減少が見られるため、今年1月に第11回新株予約権による資金調達を確保し、現状では適切に権利行使を進めている状況です。
2025年前期と比較した場合、増減としては現預金・債務ともに2億円前後の減少となっており、財務状況としては厳しいものの、将来の成長を見据えた先行投資を進めています。これに伴い、貸借対照表の改善が必要になると考えています。
事業計画(2027年3月期予想)
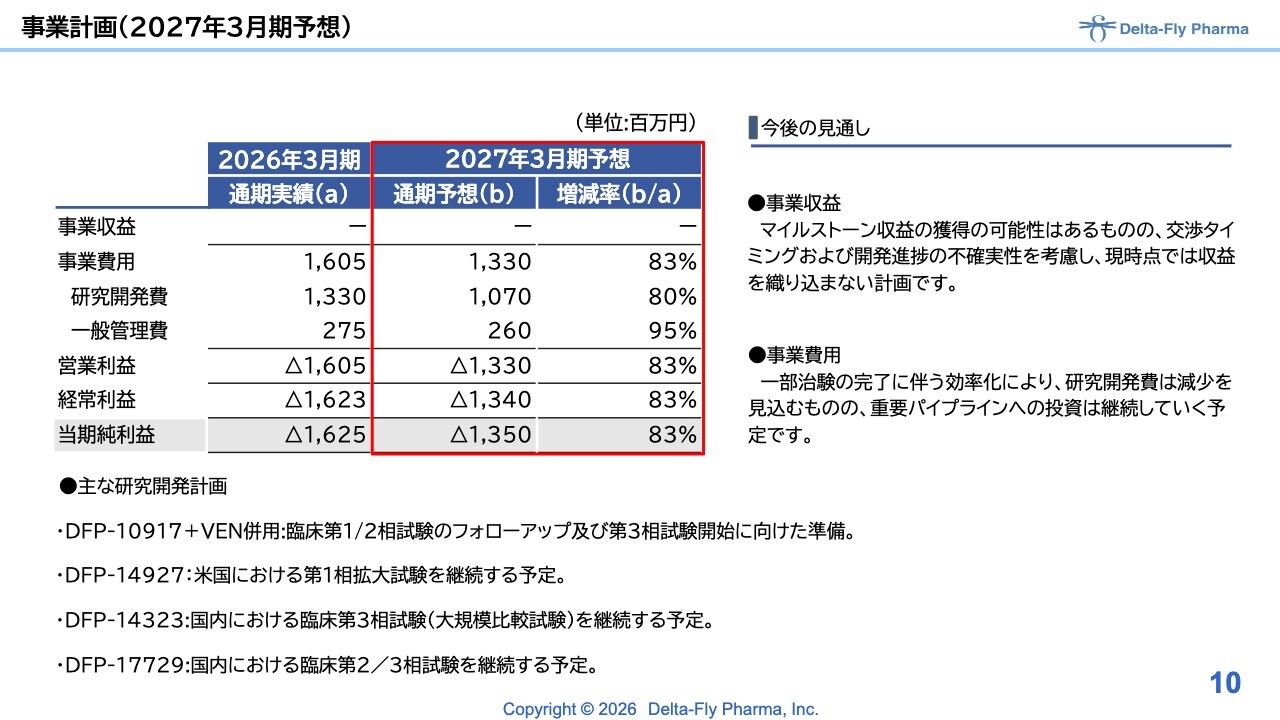
今期、2027年3月期の計画ですが、2026年3月期と比較すると、先ほど江島から説明がありましたように、「DFP-10917」の単剤についてはいったん開発が停止しています。そのため、当該開発費は発生しません。
「DFP-10917+ベネトクラクス(VEN)」の臨床第1相試験/臨床第2相試験は終了しましたが、一部フォローアップが数例残っているため、施設費用などが発生します。ただし、これらも徐々に終息する見込みであるため、「DFP-10917」関連の開発費用は圧縮できると想定しています。
「DFP-14927」については、臨床第1相試験の拡大試験にあたるため、それほど大きな費用にはならない見込みです。
国内で進めている「DFP-14323」と「DFP-17729」については、臨床第3相試験と臨床第2相試験/臨床第3相試験が進行中です。「DFP-17729」の臨床第2相試験は7月までに完了する見込みであり、その後、臨床第3相試験の準備に入る予定です。
このような研究開発費用の増減を踏まえ、全体としては、約80パーセントの経費で今期を運用できると考えています。それに伴い、営業損失は13億円強の損失を見込んでいますが、重要なパイプラインへの投資は継続して進めていきたいと考えています。
資金調達と財務状況

資金調達のハイライトです。2026年3月期は、4月に第10回新株予約権と第2回無担保社債の発行を行い、約11億5,000万円の調達を完了し、順次資金使途に充当しています。
また、1月に第11回新株予約権を発行しており、4月末時点で行使率が61パーセントとなっています。残りの権利行使についても、順次進めていく予定です。
調達資金の充当状況
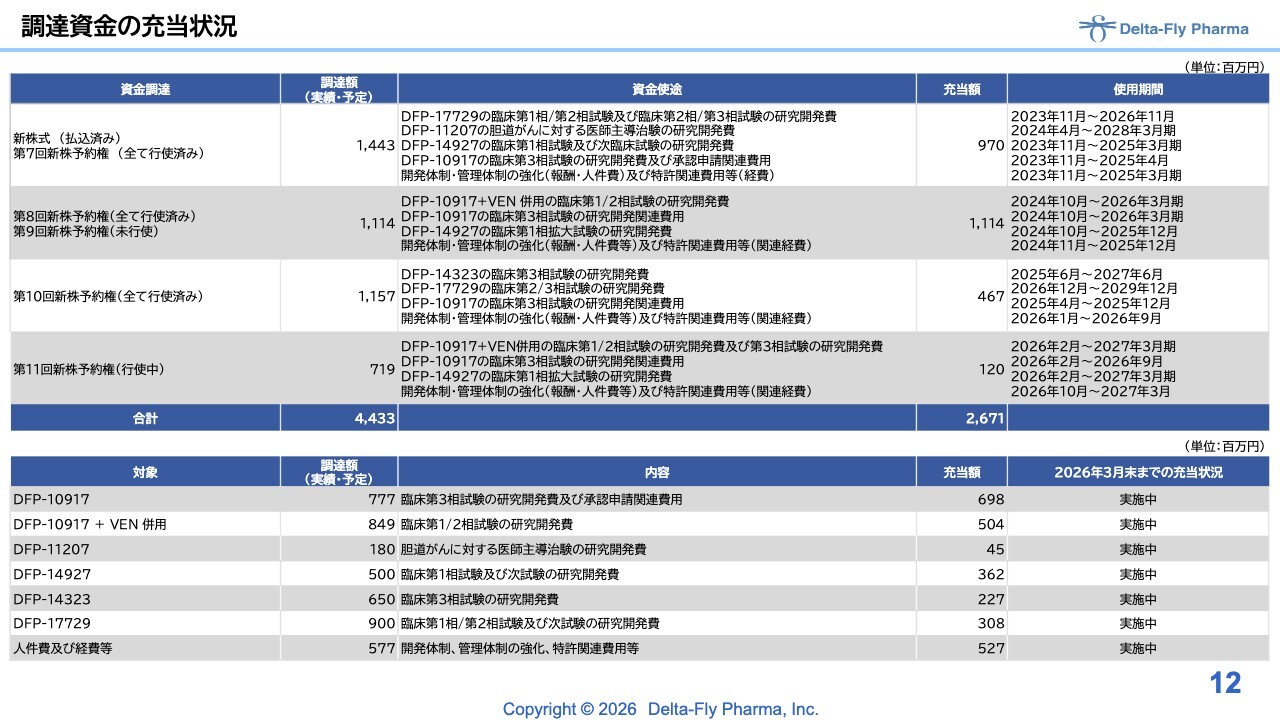
全体のこれまでの調達内容の充当状況については、スライドに記載のとおりです。
開発パイプラインの状況と今後のスケジュール
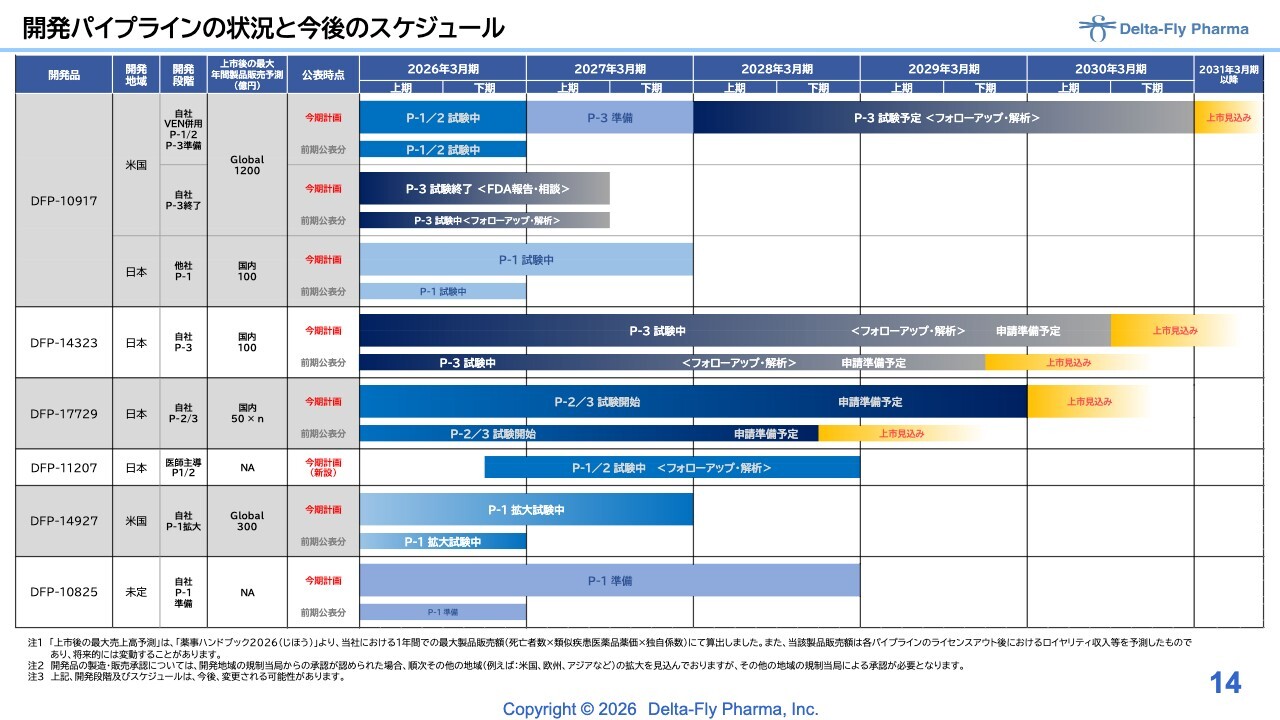
2027年3月期の全体的な計画の概略についてご説明します。各パイプラインにおける昨年公表した計画に対し、今期、新しい計画を示しています。
「DFP-10917」関連について、「ベネトクラクス(VEN)」との併用による臨床第1相試験/臨床第2相試験は、昨年の計画どおりに終了しました。今期については、臨床第3相試験の準備とフォローアップを継続し、来期から臨床第3相試験を開始する予定です。
最終的に、症例登録、フォローアップ、解析を約3年かけて進め、2031年3月期には上市に向けた申請を計画しています。
「DFP-10917」の単剤については、昨年示した計画どおりに終了しましたが、一部、計画どおりに進まなかった点がありました。そのため、FDAへの報告と相談を9月にかけて進める予定です。
日本に関しては、日本新薬による公表を基に、2027年3月期、すなわち2026年度を基準に臨床第1相試験を完了する予定となっています。
国内の「DFP-14323」については、臨床第3相試験で症例登録を行っています。一部症例の追加登録がやや遅延している部分もありますが、8ヶ月から9ヶ月ほどの延長でキャッチアップが可能と判断しており、この計画に関しては修正を加えています。
「DFP-17729」については、臨床第2相試験の開始がやや遅れたことにより、スケジュールに若干のずれが生じています。ただし、今期中に臨床第2相試験が完了し、臨床第3相試験へ進む過程にあります。
今期から来期にかけて内容の精査を行いながら、症例数の増減に応じたアダプティブ・デザインを採用した上で、臨床第3相試験の症例登録を進めていく予定です。その後、最終的な申請に向けて取り組み、2029年3月期に完了し、2030年の上市を見込んでいます。
「DFP-11207」については、今年1月に開始できる環境が整い、現在、先生方が第1症例の候補選定や症例登録の準備を進めていると聞いています。そのため、当社としては先生方からの報告を待つ段階です。
どちらにしても、臨床第1相試験/臨床第2相試験については、フォローアップと解析を約2年間で進めていく計画を立てています。
「DFP-14927」については、臨床第1相試験の拡大試験を進める予定です。
「DFP-10825」については、現状、すべてのパイプラインを同時に進めることは難しいため、臨床第1相試験の準備を延長することとしました。
以上が、各パイプラインにおける計画となっています。
ライセンス活動と財務活動の取り組み

ライセンス活動および財務活動の取り組みについての計画です。
「DFP-10917」関連を中心に、提携候補先に一定の結果を示せる状況となりつつあります。これは当然ながらNDAを締結後に可能となるものですが、現在、積極的に海外の製薬企業との交渉を進めています。今後も、新たな提携先の獲得を目指していきたいと考えています。
その他パイプラインの海外提供や共同開発も含め、連携が可能であると考えており、鋭意現状動きながら進めていきたいと考えています。
財務活動について、前期はバランスシートに大きな負担がかかりました。そのため、研究開発を推進することを目的に、ライセンス活動を進めることはもちろんですが、機動性の高い資金調達も併せて実施していきたいと考えています。
具体的には、導出活動による研究開発費の確保や新たなスポンサーの獲得を中心に進めていきます。また、ファイナンス・ストラクチャーを検討し、機動性の高い資金調達活動を実行する方針です。
以上が、今期の計画における取り組み方針となります。ご説明は以上です。
質疑応答:単剤に関する条件付き承認の意味合いと対象患者群について
質問者:単剤試験についておうかがいします。中間解析の結果を基に、単剤について条件付きで承認を行う件で米国FDAとの協議に入るとのことですが、この場合の条件付きの意味合いについて教えてください。
具体的には、対象を絞って行うという意味での条件付き承認なのか、それとも承認自体はされるものの、後から追加の第3相試験、資料や情報提供を求められるような限定的な承認といったかたちなのでしょうか?
アメリカの制度がよくわからないため、条件の意味につい
新着ログ

「医薬品」のログ